Article Text

Abstract
Introduction This post hoc pooled analysis of four real-world studies (SURE Canada, Denmark/Sweden, Switzerland and UK) aimed to characterize the use of once-weekly (OW) semaglutide, a glucagon-like peptide-1 receptor agonist (GLP-1RA), in patients with type 2 diabetes (T2D).
Research design and methods The Semaglutide Real-world Evidence (SURE) studies had a duration of ~30 weeks. Changes in glycated hemoglobin (HbA1c) and body weight (BW) were analyzed for the overall population and the following baseline subgroups: GLP-1RA-naïve/GLP-1RA switchers; body mass index <25/≥25–<30/≥30–<35/≥35 kg/m2; age <65/≥65 years; HbA1c <7%/≥7–≤8%/>8–≤9%/>9%; T2D duration <5/≥5–<10/≥10 years. Data for patients achieving treatment targets were analyzed in the overall population and the baseline HbA1c ≥7% subgroup.
Results Of 1212 patients, 960 were GLP-1RA-naïve and 252 had switched to semaglutide from another GLP-1RA. In the overall population, HbA1c was reduced from baseline to end of study (EOS) by –1.1% point and BW by –4.7 kg; changes were significant for all subgroups. There were significantly larger reductions of HbA1c and BW in GLP-1RA-naïve versus GLP-1RA switchers and larger reductions in HbA1c for patients with higher versus lower baseline HbA1c. At EOS, 52.6% of patients in the overall population achieved HbA1c <7%. No new safety concerns were identified in any of the completed SURE studies.
Conclusions In this pooled analysis, patients with T2D initiating OW semaglutide showed significant improvements from baseline to EOS in HbA1c and BW across various baseline subgroups, including patients previously treated with a GLP-1RA other than semaglutide, supporting OW semaglutide use in clinical practice.
Trail registration numbers NCT03457012; NCT03631186; NCT03648281; NCT03876015.
- glucagon-like peptide 1
- incretins
- glycated hemoglobin A
Data availability statement
Data are available on reasonable request. The data sets analysed during the current study are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
In the phase 3 Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) clinical trial program, once-weekly (OW) semaglutide, a glucagon-like peptide-1 receptor agonist, consistently demonstrated superior, clinically relevant reductions in HbA1c and body weight compared with placebo and active comparators in adults with type 2 diabetes; the safety profile of OW semaglutide was consistent with its class.
The SURE program comprises nine non-interventional, observational real-world studies investigating OW semaglutide initiation in routine clinical practice in 10 countries. To date, results from four of these studies, conducted in Canada, Denmark/Sweden, Switzerland and the UK, are available and complement the findings from the SUSTAIN clinical trials.
Significance of this study
What are the new findings?
In this pooled post hoc analysis of data from SURE Canada, Denmark/Sweden, Switzerland and UK, patients with type 2 diabetes (T2D) initiating once-weekly (OW) semaglutide showed significant improvements from baseline to end of study in HbA1c and body weight across a range of baseline characteristic subgroups, including glucagon-like peptide-1 receptor agonist (GLP-1RA) naïve/GLP-1RA switchers.
Overall, the change from baseline in HbA1c was –1.1% point, and the change from baseline in body weight was –4.7 kg.
Patients switching from another GLP-1RA to semaglutide (n=252) had significant reductions in HbA1c (–0.7% points) and body weight (–3.4 kg). In patients who were GLP-1RA naïve (n=960), these reductions were –1.2% points and –5.0 kg, respectively.
How might these results change the focus of research or clinical practice?
These results support the use of OW semaglutide in adults with T2D across multiple geographical locations in routine clinical practice, including those who have previously received a GLP-1RA other than semaglutide.
Objective
Type 2 diabetes (T2D) affects around 422 million people worldwide.1 2 The 2020 update to the consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) for T2D emphasizes preventing or delaying complications and maintaining quality of life via glycemic control.3 Other priorities include cardiovascular (CV) risk factor management and a patient-centered approach to promote self-care activities.3 Achieving glycated hemoglobin (HbA1c) targets of <7% early in the course of T2D has been shown to reduce microvascular complications.4
Many patients struggle to achieve glycemic targets, despite the availability of multiple treatment options.5 Glucagon-like peptide-1 receptor agonists (GLP-1RAs) have been demonstrated to improve glycemic control and reduce body weight and function in a glucose-dependent manner, with low risk of hypoglycemia.6 The ADA/EASD 2019 consensus statement recommends treating patients at high risk of CV disease with a GLP-1RA or a sodium–glucose cotransporter-2 inhibitor (SGLT-2i) to reduce the risk of CV events.3
Semaglutide (Novo Nordisk A/S) is a long-acting human GLP-1 analog, suitable for once-weekly (OW) dosing.7 OW subcutaneous semaglutide 0.5 mg and 1.0 mg have been approved by the US Food and Drug Administration,8 Health Canada,9 and European Medicines Agency10 to improve glycemic control in adults with T2D, in addition to diet and exercise. In the phase 3 Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) clinical trial program, OW semaglutide consistently demonstrated superior, clinically relevant reductions in HbA1c and body weight compared with placebo and active comparators across the continuum of care in T2D; its safety profile was consistent with other GLP-1RAs.11–19
Randomized controlled trials (RCTs), with their strict inclusion and exclusion criteria, may not be representative of the real-world population and clinical practice. Real-world studies can supply evidence that is complementary to the findings of RCTs, providing a more complete picture of the advantages and disadvantages of medications used in routine clinical practice.20
The Semaglutide Real-world Evidence (SURE) program comprises nine observational real-world studies investigating OW semaglutide initiation in routine clinical practice in 10 countries: Canada (CA), Denmark/Sweden (DK/SE), France, Germany, Italy, Spain, The Netherlands, Switzerland (CH), and the UK. Each study has been registered separately, and all are similar in design, but their patient populations vary, depending on the respective countries’ interest in specific subgroup analyses.
The results from the first four individual SURE studies to report (CA (n=452),21 DK/SE (n=331),22 CH (n=214)23 and UK (n=215))24 showed that patients receiving OW semaglutide experienced statistically and clinically significant improvements in glycemic control and reduction in body weight. Because of the impact of the coronavirus (COVID-19) pandemic on the timelines of the incomplete SURE studies, as well as the resultant changes to protocols, it was decided to perform a pooled analysis only of the first four available studies. The date of the first patient, first visit for SURE Canada, the first study in the SURE program, was March 2018, and the last patient last visit for the most recent SURE study in this analysis, SURE UK, took place in August 2020.
This pooled post hoc analysis of the first four SURE studies aimed to characterize the use of OW semaglutide in diverse patient populations, with greater statistical power allowing assessment in patient subgroups. The main subgroups of interest are GLP-1RA-naïve patients (ie, those not receiving another GLP-1RA ≤12 weeks prior to semaglutide initiation) and patients who switched to OW semaglutide from another GLP-1RA. The patient data were also subgrouped by baseline body mass index (BMI), baseline age, baseline HbA1c level and duration of T2D. In addition, the proportions of patients achieving glycemic targets and weight-loss responses were evaluated.
Methods
Study design of SURE studies
The study design and endpoints were similar for all four SURE studies and have been reported elsewhere.21–24 The participating clinics were selected in close collaboration with the Novo Nordisk affiliates in the various countries to ensure representativeness of the local populations. The study duration was approximately 30 weeks. The SURE UK study, however, allowed patients to attend the end-of-study (EOS) visit up to week 52, because of the COVID-19 pandemic. Data for the 34 patients (18.6%) who attended the EOS visit in the extended period of the SURE UK study were included in the primary analysis for SURE UK and are therefore in the present analysis as well. The SURE CA, DK/SE and CH studies were completed before the pandemic. Patients ≥18 years of age with T2D who had ≥1 documented HbA1c value ≤12 weeks before semaglutide initiation were enrolled. Patients were retained in the full analyses set (FAS) if they provided informed consent and initiated semaglutide. Semaglutide and other antihyperglycemic drugs were prescribed at the physician’s discretion. Treatment discontinuation was allowed at any time during the study at the physician’s discretion. The studies were conducted in compliance with the Declaration of Helsinki25 and the Guidelines for Good Pharmacoepidemiology Practices.26 Patients provided informed consent before the commencement of any study-related activities.
The primary endpoint was the change from baseline to EOS in HbA1c. Secondary supportive endpoints included: change from baseline to EOS in body weight (kg and %) and waist circumference (cm); proportion of patients achieving HbA1c <7%, weight loss ≥5%, and a composite endpoint of HbA1c reduction ≥1% point and weight loss ≥3%; patient-reported outcomes (Diabetes Treatment Satisfaction Questionnaire status version (DTSQs), DTSQ change version (DTSQc) and Short-Form 36 Health Survey V.2).27 28
Only serious adverse drug reactions (SADRs), fatal events, accidental pregnancies, adverse events (AEs) in fetus or newborn infants and discontinuation due to SADRs were systematically recorded by site physicians at each visit. All other AE information was collected if reported voluntarily by the physicians. All episodes of patient-reported hypoglycemia and/or severe or documented hypoglycemia (blood glucose ≤3.9 mmol/L or >3.9 mmol/L in conjunction with symptoms) were also to be recorded.
Post hoc analysis
A random coefficient-adjusted mixed model for repeated measurements was used for all assessments of the FAS, including the entire time period when patients were considered to be in the study, regardless of semaglutide treatment status. The analysis included all patients in the FAS with at least one postbaseline HbA1c measurement. Components of the model included time of measurement (number of days from baseline; continuous variable), a time component (t (squared)), to account for any deviations from linearity in time; baseline HbA1c (continuous variable); preinitiation use of GLP-1RA (yes/no; not included in subgroup analyses based on GLP-1RA-naïve/switcher); preinitiation use of dipeptidyl peptidase-4 inhibitor (DPP-4i; yes/no); preinitiation use of insulin (yes/no); number of oral antihyperglycemic drugs used preinitiation (0–1/2+); T2D duration (continuous); age (continuous); BMI (continuous); and sex; with random intercept and random coefficient for time. An unstructured covariance matrix was used to describe the variability between random effects. From this model, the estimated difference between HbA1c at week 30 versus baseline at week 0 is presented, together with the associated two-sided 95% CI and adjusted two-sided p value. To test for interaction of subgroups, the subgroup being evaluated was added as a covariate in the main model.
HbA1c and body weight were analyzed for the overall pooled population (prespecified) and in the following baseline subgroups (post hoc): GLP-1RA-naïve (no GLP-1RA use reported during the 12 weeks prior to baseline) and GLP-1RA switchers (baseline GLP-1RA users who discontinued within 4 weeks of initiating semaglutide, allowing for a smooth switch); baseline BMI <25, ≥25–<30, ≥30–<35 and ≥35 kg/m2; age <65 years and ≥65 years; baseline HbA1c <7%, ≥7–≤8%, >8–≤9% and >9%; T2D duration <5, ≥5–<10, ≥10 years; and reason for the initiation of semaglutide. The proportions of patients in the overall pooled population achieving the following treatment targets and responses at EOS were analyzed: HbA1c <7%, weight loss ≥3%, weight loss ≥5%, weight loss ≥10%, and a composite endpoint of HbA1c reduction ≥1% point and weight loss ≥3%. In addition, the proportions of patients achieving HbA1c <7% and the composite endpoint in the subgroup of patients with a baseline HbA1c ≥7% were also analyzed. Dose at EOS and severe or documented hypoglycemic episodes were analyzed using the effectiveness analysis set (EAS), which included all patients in the FAS who completed the study on treatment with semaglutide; all other endpoints were analyzed using the FAS.
Results
Patient disposition and baseline characteristics
Across the four SURE studies, 1212 patients were included in this post hoc analysis (FAS), with a mean semaglutide treatment duration within the studies of 30.8±9.6 weeks. In the EAS, there were 984 patients. The baseline characteristics were typical of real-world practice. Mean age was 60.1 years, mean diabetes duration was 12.2 years. The majority (91.0%) of the study population was white. Mean HbA1c was 8.1%; 231 (19.1%) patients had a baseline HbA1c <7.0%. Overall, 252 patients switched to semaglutide from another GLP-1RA, whereas 960 were GLP-1RA naïve (table 1). Six patients in the GLP-1RA switcher group did not have a stop date for the previous GLP-1RA registered within the first 4 weeks of initiating semaglutide. These patients were included in the analysis under the assumption that there is no stop date because these data are missing rather than these patients were receiving both GLP-1RAs simultaneously. Mean baseline HbA1c was greater in GLP-1RA-naïve patients (8.2%) versus GLP-1RA switchers (7.8%). GLP-1RA switchers had a longer diabetes duration (13.7 years) than GLP-1RA-naïve patients (11.8 years). Slightly greater proportions of GLP-1RA switchers had comorbid conditions than GLP-1RA-naïve patients, with the exception of diabetic retinopathy, diabetic neuropathy, and heart failure (table 1). Baseline characteristics for other subgroups investigated are included in online supplemental tables 1–4.
Supplemental material
Demographics and baseline characteristics of patients
Overall, 937 (77.3%) patients were initiated on a 0.25 mg dose. The majority of GLP-1RA-naïve patients were prescribed a starting semaglutide dose of 0.25 mg, whereas approximately half of the patients in the GLP-1RA switcher group started on a 0.5 mg or 1.0 mg dose, compared with only about 16% of GLP-1RA-naïve patients (online supplemental tables 5–8). For the majority of patients (1020 (84.2%)), one of the reasons for initiating OW semaglutide was to improve glycemic control, with weight reduction as a secondary reason. The rationale for initiating semaglutide was broadly similar for both GLP-1RA-naïve patients and GLP-1RA switchers (table 1).
Overall, 941 (77.6%) patients were taking metformin at baseline, 499 (41.2%) were on an SGLT-2i and 421 (34.7%) on basal insulin. At baseline, 201 patients were on a DPP-4i, of which 131 switched to semaglutide and 70 had semaglutide added on to a DPP-4i (table 2). Medications at baseline for the other subgroups investigated are shown in online supplemental tables 5–8.
Antihyperglycemic medication at baseline in the overall population and by GLP-1RA status
HbA1c
The change from baseline in HbA1c was –1.1% point. The change overall and changes from baseline to week 30 for all subgroups tested were significant (p<0.0001). However, the difference in change between subgroups (test for interaction) was only significant for GLP-1RA-naïve patients versus GLP-1RA switchers (see later section) and baseline HbA1c subgroups, while numerical differences were seen in several other subgroups (figure 1A). HbA1c reductions were significantly greater in patients in the >9% HbA1c group (–2.5% point) versus the <7%, ≥7–≤8% and >8–≤9% baseline HbA1c groups (–0.2% point, –0.7% point and –1.1% point, respectively; interaction p=0.0209). When stratified by reason to initiate semaglutide, the reductions in HbA1c were similar and similar to the overall reduction of –1.1% point (online supplemental figure 1).

(A) Change in HbA1c from baseline to EOS in overall population and subgroups; (B) change in body weight from baseline to EOS in overall population and subgroups. Data are from the full analysis set, in-study period that represents the time period during which patients are considered to be in the study, regardless of semaglutide treatment status. Response was analyzed using baseline T2D duration, age, BMI, time, time-squared, preinitiation use of DPP-4i, preinitiation use of insulin, preinitiation use of GLP-1RAs, GLP-1RA (except in ‘GLP-1RA experience’ subgroups), number of OADs used preinitiation (0–1/2+) and sex with random intercept and random time coefficient (slope). (A) All p values for change from baseline to week 30 are significant at <0.0001. Interaction p value for difference in change between subgroups: *p=0.0003; †0.0209; ‡0.9354; ‖0.1944; §0.3504. (B) All p values for change from baseline to week 30 are significant at <0.0001 except p=0.0092 for baseline BMI of 25 kg/m2. Interaction p value for difference in change between subgroups: *p<0.0001; †0.8730; ‡0.5791; §0.8419; ‖0.7569. BMI, body mass index; DPP-4i, dipeptidyl peptidase-4 inhibitor; EOS, end of study; GLP-1RA, glucagon-like peptide-1 receptor agonist; HbA1c, glycated hemoglobin; OAD, oral antihyperglycemic drug; T2D, type 2 diabetes.
Body weight
Overall, the change from baseline in body weight was –4.7 kg. The change from baseline to EOS was significant (p<0.01) for all subgroups tested. Numerical differences were seen in several subgroups (figure 1B) with the only significant difference between subgroups (test for interaction) for GLP-1RA-naïve (–5.0 kg) patients versus GLP-1RA switchers (see following section).
Treatment responses among patients switching or not switching from another incretin agent (GLP-1RA or DPP-4i)
HbA1c reductions were significantly greater in patients who were GLP-1RA-naïve versus those who switched from another GLP-1RA to semaglutide (–1.2% point vs –0.7% point, respectively; interaction p=0.0003) (figure 1A). The change in body weight from baseline to EOS also differed significantly between these groups (interaction p<0.0001); –5.0 kg on average for GLP-1RA-naïve patients versus −3.4 kg for GLP-1RA switchers.
HbA1c reductions were similar for patients switching from a DPP-4i to semaglutide at baseline (n=123) and those who received semaglutide in addition to a DPP-4i (n=70) (–1.3% for both, interaction p=0.3594). The body weight reductions in patients switching from a DPP-4i to semaglutide (n=131) at baseline (–5.6 kg) were similar to those in patients who initiated semaglutide in addition to a DPP-4i (n=70) (–4.4 kg, interaction p=0.4834).
Treatment targets and composite endpoints
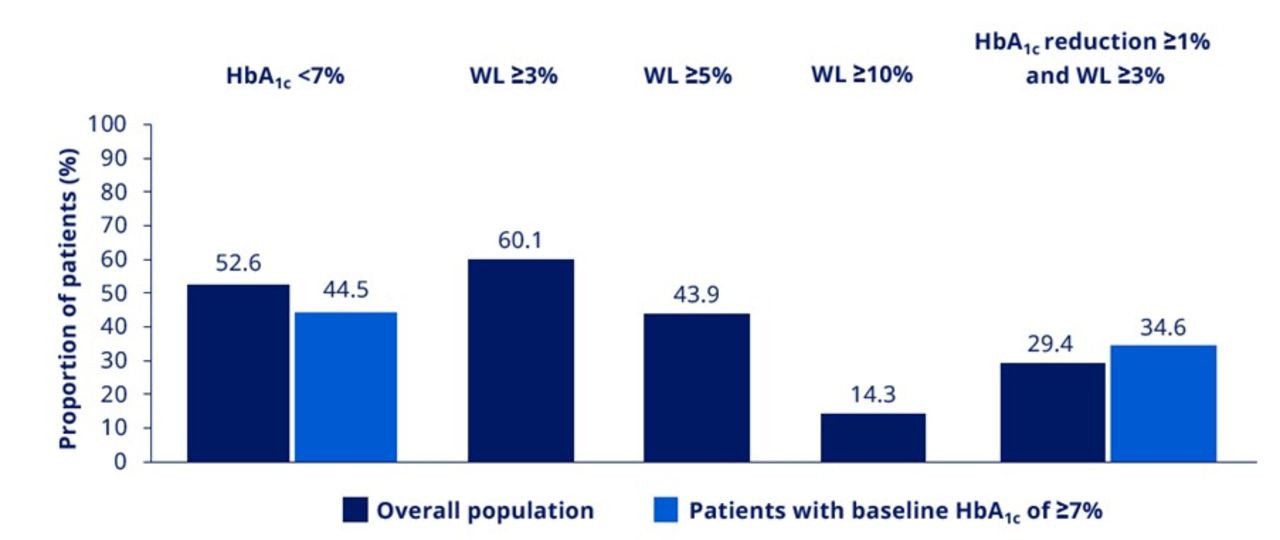
At EOS, 531 (52.6%) patients in the overall pooled population and 365 (44.5%) patients with a baseline HbA1c ≥7% achieved HbA1c <7%. At EOS, 609 (60.1%), 445 (43.9%) and 145 (14.3%) patients in the overall pooled population achieved weight loss ≥3%, ≥5% and ≥10%, respectively (figure 2). At EOS, 297 (29.4%) patients in the overall population and 283 (34.6%) patients with a baseline HbA1c ≥7% achieved the composite endpoint of an HbA1c reduction ≥1% point and weight loss ≥3% (figure 2).
{kind=link}
{kind=link}
Proportions of patients achieving treatment targets at EOS. Data are based on the full analysis set. EOS, end of study; HbA1c, glycated hemoglobin; WL, weight loss.
Semaglutide dose at EOS
The mean dose of semaglutide at EOS in the EAS was 0.8±0.30 mg. At EOS, 7 (0.7%) patients were receiving a semaglutide OW dose <0.25 mg; 109 (11.1%) a 0.25 mg dose; 3 (0.3%) were receiving a dose between 0.25 and 0.5 mg; 274 (27.8%) were receiving a 0.5 mg dose; and 13 (1.3%) were receiving a dose between 0.5 and 1.0 mg. The majority of patients (576 (58.5%)) were taking the 1.0 mg dose and 2 (0.2%) were taking a dose >1.0 mg. The use of doses <0.25 mg and between 0.25 mg and 0.5 mg, between 0.5 and 1.0 mg, and over 1.0 mg is off-label and occurred because of the studies’ non-interventional nature.
Safety
No new safety concerns were identified with OW semaglutide in any of the four completed SURE studies (systematically collected safety information reported in online supplemental table 10; voluntarily reported adverse events reported in online supplemental table 11). In the pooled analysis of 1212 patients, 115 (9.5%) patients discontinued treatment, of which 73 (63.5%) did so because of unacceptable gastrointestinal tolerability. Adverse events led to premature discontinuation of treatment in 44 (3.6%) patients (online supplemental table 11). In the EAS, there were 69 patients (6.0%) who experienced severe or documented hypoglycemic episodes (online supplemental table 10). There were two patients who experienced severe hypoglycemia, both of whom were receiving concomitant insulin therapy.
Discussion
In this pooled analysis of real-world data from SURE CA, DK/SE, CH and UK, patients treated with OW semaglutide experienced clinically relevant and statistically significant reductions in HbA1c and body weight in the overall population and in subgroups stratified by various baseline characteristics, including prior treatment with a GLP-1RA other than semaglutide. These results are in line with the findings of the SUSTAIN phase 3 RCTs. In the overall population, more than 50% of patients achieved the ADA-recommended HbA1c target <7%, and 44% of patients achieved a weight loss ≥5% by EOS.
The SURE studies enabled an assessment of the effects of switching from another GLP-1RA to semaglutide, which has not yet been explored in RCTs. Patients switching from another GLP-1RA experienced statistically and clinically significant reductions in HbA1c (–0.7% point) and body weight (–3.4 kg), despite switching from an agent of the same class. This finding is consistent with the EXPERT study of a US electronic medical record database that showed that patients switching from another GLP-1RA to OW semaglutide had significant and sustained reductions in HbA1c and body weight.29 Similar findings were also reported in the retrospective Switch-to-Semaglutide Study.30 In the retrospective study by Goncalves and Bell,31 40 patients in an endocrine practice in Canada who had switched from liraglutide to OW semaglutide experienced an HbA1c reduction of 0.8% point and a body weight reduction of 4.6 kg following the switch. The REALISE-DM study, a retrospective chart review of 164 patients with T2D in an endocrine practice in Canada, demonstrated that switching to OW semaglutide from dulaglutide or liraglutide resulted in a further significant HbA1c reduction of 0.7% and body weight loss of 1.6 kg at 6 months.32 These observations are consistent with those from the SUSTAIN 7 head-to-head RCT, which showed that semaglutide was superior to dulaglutide in reducing HbA1c and body weight.16
Because of the non-interventional nature of the SURE studies, semaglutide was added to existing DPP-4i treatment in a small proportion of patients. This prescribing practice is not recommended in treatment guidelines3; however, it is important to confirm that this practice does not impact the change in HbA1c or body weight. In this pooled analysis, patients who had switched from a DPP-4i at baseline to OW semaglutide had similar reductions in HbA1c to patients who remained on their DDP-4i therapy after initiation of semaglutide. This indicates that remaining on DPP-4i treatment after initiating semaglutide had no additional benefit on glycemic control and supports the recommended withdrawal of DPP-4i treatment after initiation of semaglutide. The specific reasons for prescribing semaglutide as an add-on to DPP-4i therapy were not recorded; possibly the prescribers’ intention was to keep these patients on a drug with known efficacy and tolerability, if semaglutide needed to be discontinued, with the ultimate aim of discontinuing the DPP-4i once the semaglutide dose had been increased to a therapeutic level and tolerability had been established.
The HbA1c reductions observed in the SURE studies were lower than those in the SUSTAIN RCTs. The protocol used for the SURE studies differed from the one for the SUSTAIN RCTs in regard to the study design, initiation and usage of semaglutide throughout the studies and data collection. The difference in inclusion/exclusion criteria is likely to have contributed to the lower HbA1c reduction in the SURE studies versus the SUSTAIN trials (online supplemental table 9). For example, all the SUSTAIN trials included baseline HbA1c ≥7.0% or ≥7.5% with an upper limit of 10.0%, 10.5% or 11.0% as part of the inclusion criteria,11–19 whereas the SURE studies had no such criteria. The difference in the level of treatment adherence may also have impacted the results. Patients in routine clinical practice generally have poorer medication adherence than those enrolled in an RCT: this has been observed in other real-world studies.33 Another key difference is that the dose escalation of semaglutide and selection of maintenance doses were prespecified in the protocols of the trials in the SUSTAIN program, whereas in the SURE studies, the treating physicians determined how the dose would be escalated and which maintenance dose should be used. The lower baseline HbA1c of patients in the SURE studies (8.1%) compared with the SUSTAIN program (8.0%–8.4%), and the inclusion in the SURE program of patients with baseline HbA1c <7%, may have contributed to the comparatively lower reduction in HbA1c from baseline to EOS.11–19 Mean baseline body weight in the SURE studies (101.5 kg) was higher than in the SUSTAIN trials (89.2–96.9 kg). The body weight reduction, however, was comparable between the SURE studies (–4.7 kg) and the SUSTAIN trials (–3.5 kg to –6.4 kg).11–19
The proportion of patients discontinuing treatment due to an AE in the SURE studies (9.5%) was lower than was observed with OW semaglutide 1.0 mg in the SUSTAIN clinical trial program (≤15%).11–19 34 This highlights that OW semaglutide is well tolerated in real-world practice. This discontinuation rate was also lower than the rate observed in the retrospective observational SPARE study (17%), which included data from 937 GLP-1RA-naïve patients with T2D.35 This difference may be due to variations in dosing practices. In the SUSTAIN clinical trial program, patients were required to follow a clear dosing schedule during the study period; in contrast, in real-world practice, dosing schedules may differ depending on patient needs: to manage GI side effects, for example, the escalation strategy during initiation or maintenance dosing (which may include dose skipping) will be tailored to the individual. The initial recommended dose for semaglutide is 0.25 mg, before escalating to a maintenance dose of 0.5 mg after 4 weeks and to 1.0 mg after a further 4 weeks, if needed.11–19 However, in the SURE studies, approximately 77% of patients were initiated on a dose of 0.25 mg and, by EOS, the 576 (58.5%) patients remaining on treatment were receiving a 1.0 mg dose. While doses other than 0.5 and 1.0 mg/week are off-label, such doses were being used by 134 (14%) of patients still on treatment in the study (because of the non-interventional nature of the study), which is an indication that patients and physicians were tailoring the dose to individual requirements.
The SURE studies provide information on patients with T2D with a wide range of baseline characteristics in routine clinical practice in diverse locations. For example, in SURE UK, the mean BMI and HbA1c of the patient population at baseline were slightly higher than for patients in SURE Canada, SURE Denmark/Sweden and SURE Switzerland. No new safety concerns were identified in the four SURE studies, highlighting that OW semaglutide is well tolerated in real-world practice.
The main limitation of the SURE studies relates to the one-armed observational design and lack of a comparator. In the absence of a randomized comparator group, we cannot rule out the impact of other factors nor directly infer that the estimated changes in the outcomes are causal effects of study treatment. Regression to the mean may also have contributed to the observed changes in the outcomes. Other limitations relate to the observational nature of the studies, in that data were collected as part of routine clinical practice rather than through mandatory assessments at prespecified time points, which may have affected the robustness and completeness of the data.
Conclusion
In a pooled analysis of the SURE Canada, Denmark/Sweden, Switzerland and UK studies, patients with T2D initiating OW semaglutide experienced significant improvements from baseline to week 30 in HbA1c and body weight, both in the overall pooled population and across subgroups characterized by various baseline characteristics, including the subgroup who switched from a GLP-1RA other than semaglutide. At EOS, over half of the patients in the overall pooled population had an HbA1c <7%, and over 40% with a baseline HbA1c ≥7% had achieved an HbA1c <7%. Safety data collected during the studies showed no new safety concerns with semaglutide, and the benefit–risk balance remains positive. The results support the use of OW semaglutide in adults with T2D in routine clinical practice across multiple geographical locations.
Data availability statement
Data are available on reasonable request. The data sets analysed during the current study are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The SURE studies included in this analysis were conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Pharmacoepidemiology Practices. All patients provided their prior, informed consent for participation in this study. Study materials were approved by institutional review boards or other appropriate local bodies. The SURE Canada study materials were approved by Schulman IRB (reference number: 201708875). The SURE Denmark/Sweden study materials were approved by Regionala Etikprövningsnämnden i Stockholm (reference number: 2018/1341-31/2), ethical approval was not needed for Denmark. The SURE Switzerland study materials were reviewed and approved by the Ethikkommission der Nordwest und Zentralschweiz EKNZ, Wissenschaftliches Sekretariat (reference ID: 2018-01028). The SURE UK study materials were approved by the South West—Central Bristol Research Ethics Committee (reference number: 19/SW/0048).
Acknowledgments
We would like to thank all the participants, investigators, and trial-site staff, as well as Andreas Ross Kirk (Novo Nordisk, Søborg, Denmark) and Mohd Tariq (Novo Nordisk, Bangalore, India) for their review and input into the manuscript, and Priya Talluri and Catherine Starling (AXON Communications) for medical writing and editorial assistance (funded by Novo Nordisk A/S). As the guarantor of this manuscript, Andrei-Mircea Catarig (Novo Nordisk A/S, Søborg, Denmark) takes full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors contributed to the writing and editing of the manuscript, and JF-Y and A-MC supervised the study. JF-Y, SC, AC, NRE, PH, STK, TS and GR contributed to the study investigation. JF-Y, A-MC and NRE contributed to the conceptualisation of the study. A-MC, AC and PH contributed to the study methodology. A-MC, NRE, UE and TS contributed to the curation and analysis of the data.
Funding The SURE programme was funded by Novo Nordisk A/S.
Competing interests J-FY reports receiving grants from Novo Nordisk during the conduct of the study; grants and personal fees from Novo Nordisk, Eli Lilly, Boehringer Ingelheim, Merck, Janssen, AstraZeneca, and Sanofi, all outside the submitted work. UB reports personal fees for participation in a scientific advisory board from Novo Nordisk, outside the submitted work. A-MC, AC and UE are employees of Novo Nordisk, and A-MC and UE own stock in the company. SC reports consultancy payment for Novo Nordisk (paid to his employer). NRE reports payment for lecturing and reimbursement for participation in scientific advisory boards from Novo Nordisk (paid to her employer), outside the submitted work. PH reports personal fees from AstraZeneca, Eli Lilly, and Novo Nordisk, outside the submitted work. STK reports grants and personal fees for lectures and/or consultancy from AstraZeneca, Boehringer Ingelheim, and Novo Nordisk, and personal fees for lectures and/or consultancy from MSD, Mundipharma and Sanofi, outside of the submitted work. JL reports compensation for clinical trial research, personal fees from continuing medical education events, outside the submitted work. TS reports grants from Abbott and Novo Nordisk outside the submitted work. BS reports fees for advisory board meetings and lectures from Novo Nordisk. GR reports research funding from Novo Nordisk.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.