Article Text

Abstract
Introduction To investigate the role of CD36 (fatty acid translocation enzyme) in the myocardial ischemia reperfusion (IR) injury in diabetes with ischemic postconditioning (IPostC).
Research design and methods Adult male Sprague-Dawley rats received streptozotocin treatment to establish type 1 diabetic model. After 8 weeks, diabetic rats were subjected to myocardial IR and IPostC with or without sulfo-N-succinimidyl oleate (SSO, an inhibitor of CD36) intervention.
Results Diabetic rats showed the upregulation of myocardial CD36 expression and the increase in free fatty acid (FA) and triglycerides (TG) level and FA β oxidation (FAO). The cardioprotection of IPostC was compromised in diabetic rats with myocardial IR as evidenced by increased myocardial infarct size and plasma levels of lactate dehydrogenase (LDH), creatine kinase MB isoenzyme (CK-MB), and cardiac troponin Ⅰ (cTn-I), but not in non-diabetic rats with myocardial IR. SSO significantly decreased the levels of plasma LDH, CK-MB, cTn-I, free FA, and the levels of myocardial malondialdehyde, 8-isoprostane, FA, TG, and CD36 expression, and significantly increased the levels of myocardial glutathione peroxidase, total glutathione/oxidized glutathione, FAO, peroxisome proliferator activated receptor alpha, pyruvate dehydrogenase kinase 4, and the early (E) and late (A) diastolic filling ratio of heart in diabetic rats with IR and IPostC. However, no significant differences were observed in myocardial infarct size, heart rate, ejection fraction, fractional shorting, and dp/dtmax.
Conclusions CD36 downregulation partially attenuated myocardial IR injury in diabetic rats with IPostC via ameliorating FA metabolism and oxidative stress.
- CD36 antigens
- myocardium
- ischemia
- diabetes mellitus, type 1
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Fatty acid (FA) metabolic dysfunction impaired diabetic myocardial flexibility, which was associated with ischemia reperfusion (IR) injury susceptivity and ischemic postconditioning (IPostC) hyporesponsivity.
WHAT THIS STUDY ADDS
Myocardial CD36 overexpression was accompanied by the attenuation of IPostC cardioprotection in diabetic rats.
CD36 downregulation partially attenuated myocardial IR injury in diabetic rats with IPostC via ameliorating FA metabolism and oxidative stress.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
CD36 would be significant for advancing the development of a new treatment in patients with ischemic heart disease complicated with diabetes.
Introduction
Diabetes mellitus (DM) renders the heart more vulnerable to ischemia reperfusion (IR) injury1 and compromises the cardioprotection of ischemic postconditioning (IPostC).2 However, the mechanisms are still not well understood.
Cardiomyocytes derive most energy from fatty acids (FAs). Regulation of FA uptake and oxidation is an important contributor to myocardial energy metabolism. Previous research found that the FA translocation enzyme (also named as CD36) accounted for 53% of myocardial total FA uptake.3 The induced myocardial CD36 expression was responsible for increased FA uptake, thereby causing lipotoxicity in the heart,4 5 which was considered as an important component of the pathogenesis of cardiac complications of DM.6 It is noteworthy that CD36 downregulation by the empagliflozin treatment provided protective effects against the development of cardiometabolic diseases in Zucker diabetic fatty rats.7 Silencing CD36 or GW9662 treatment could protect rats against diabetic cardiomyopathy via CD36 inhibition.4 Besides, elective CD36 ligand pretreatment could reduce myocardial infarct size, FA uptake and preserve hemodynamics in mice subjected to myocardial IR but have no effect in CD36-deficient mice.8 9 These data suggested that CD36-regulated FA metabolism might play an important role in diabetic myocardial IR.
As reported, pre-incubated primary cardiomyocytes with sulfo-N-succinimidyl oleate (SSO, anti-CD36-specific binding molecule) enhanced basal glucose uptake and prevented triacylglycerol accumulation and contractile dysfunction.10 And, CD36 downregulation by SSO effectively attenuated hypoxia/reoxygenation injury in the isolated diabetic heart by significantly decreased FA uptake, conversion and oxidation.11 Our previous study showed that diabetes induced a significant increase in cardiac CD36 expression, which was associated with reduced sevo-postC cardioprotection.12 We here hypothesized that inhibition of CD36 by SSO could attenuate myocardial IR injury in diabetes with IPostC.
Materials and methods
Animals
Male Sprague-Dawley rats (250±10 g body weight, aged 8 weeks) were supplied by the Laboratory Animal Service Center of Wuhan University and had free access to food and water before the start of the experiments. All rats were maintained under specific pathogen-free conditions and were housed in a laboratory room with a 12-hour dark and 12-hour light cycle, a room temperature of 22°C–24°C, and a humidity of 50%–60%. Water intake and food consumption were recorded daily, while blood glucose and body weight were monitored weekly.
Diabetic model of rats
As previously described,13 diabetes was induced by a single tail vein injection of streptozotocin (Sigma-Aldrich, S0130) at the dose of 60 mg/kg body weight in 0.1 M citrate buffer (pH 4.5) or citrate buffer alone as the control. After 3 days injection, blood glucose was measured using a glucose meter (LifeScan) and rats with fasting blood glucose levels over 16.7 mM at least three times were considered diabetes.
Myocardial IPostC model in vivo and SSO treatment
Eight weeks after the onset of diabetes, a well-established model of myocardial IPostC in vivo was used.13 Rats, after induction by intraperitoneal injection of 1% pentobarbital sodium (60 mg/kg), were intubated with an endotracheal tube and connected to a rodent ventilator with a tidal volume of 1.0 mL/100 mg body weight and 70 breaths/min. Anesthesia was maintained with inhalational sevoflurane at a concentration from 2.0% to 3.0% until the end of the experiments. During the experiment, all rats were kept warm by a lamp and the vital signs were measured. Cardiac ischemia was achieved by occluding the left anterior descending (LAD) coronary artery by 6–0 silk suture using a snare occluder for 0.5 hours. Ischemia was verified by myocardial discoloration of the ischemic zone and elevation of ST-segment in limb lead II. IPostC was performed by three cycles of 10 s of reperfusion and 10 s of ischemia after myocardial ischemic, then followed by reperfusion for 2 hours. SSO was obtained from Santa Cruz Biotechnology (sc-208408) and freshly dissolved in pure dimethyl sulfoxide (DMSO) to obtain a concentration of 4 mM, then freshly diluted with normal saline to prepare a 0.5 mM for working solution. SSO was applied intravenously at a rate of 0.2 mL/hour from 4 min before ischemia to the end of reperfusion in diabetic rats based on the previous study11 and our preliminary experiments. Plasma was extracted from blood samples and stored at −80°C until assay. The ventricular tissue was removed and immediately frozen in liquid nitrogen and stored at −80°C for further analysis.
Echocardiographic assessment of cardiac function
Transthoracic echocardiography was performed non-invasively at experiment termination with Vevo 770 high-resolution imaging system equipped with a 17.0 MHz transducer (RMV-716, Visual Sonics), and left ventricular (LV) dimensions and LV diastolic and systolic functions were assessed by M-mode and Doppler echocardiography. The ratio of the peak velocity of early (E) and late (A) diastolic filling (E/A), left ventricular internal dimensions at end-systole and diastole (LVIDs and LVIDd), LV end-diastolic volume and end-systolic volume (LVVd and LVVs) were monitored. Ejection fraction (%)=(LVVd–LVVs)/LVVd×100%, fractional shorting (%)=(LVIDd–LVIDs)/LVIDd×100%. All derived measures by echocardiography were obtained by averaging the readings of three consecutive beats.
Hemodynamic monitoring
The hemodynamic monitoring of diabetic animals was performed as we described.13 Briefly, a saline-filled catheter was inserted into the left ventricle via an incision on the right common carotid artery and then connected to a pressure transducer (SCW Medicath, DPT-248). The left ventricular systolic pressure, left ventricular maximum rate of increase of left ventricular developed pressure (dp/dtmax), maximum rate of decrease of left ventricular developed pressure (dp/dtmin), and heart rate were monitored (Bene View T5, Xi’an Jutian Medical Equipment, China) at 10 min before ischemia (baseline) and 2 hours after reperfusion.
Determination of myocardial infarct size
After the reperfusion was finished, rats were sacrificed by an overdose of anesthetic sevoflurane. Then, the LAD was reoccluded and cannulated just distal to the occlusion site. 10 mL of saline and 10 mL of 0.3% Evans blue dye were injected with equal pressure into the aorta root, respectively. Then, hearts were immediately fibrillated, removed, weighed, and frozen at −20°C for 20 min, underwent horizontal long axis slicing at a thickness of 1–2 mm, incubated at 37°C for 20–30 min in 1% 2,3,5-triphenyl tetrazolium chloride (TTC) staining (Sigma, USA) in 0.1 mol/L phosphate buffer adjusted to pH 7.4 and scanned with a digital camera. ImageJ V.1.47 (National Institutes of Health) was used to measure and analyze the infarct size. The unstained region by Evans blue dye was considered as the area at risk (AAR). The unstained region of TTC was identified as the area of infarction. Infarct size was expressed as a percentage of the AAR.
Measurement of fatty acid β oxidation rate
The fatty acid β oxidation (FAO) rate of cardiac tissue was measured using the Fatty Acid β Oxidation Rate Assay kit (HPBIO-JM10629) from HePeng (Shanghai) Biotech (Shanghai, China) according to the manufacturer’s specifications. Briefly, cardiac tissues were homogenized in a lysis buffer (100 mg tissues/1 mL) at 0°C. After incubated at 4°C for 10 min, the homogenate was centrifuged at 1000 g for 5 min at 4°C to obtain the supernatant which was centrifuged at 12 000 g for 10 min at 4°C to obtain the pellet. The pellet was resuspended with 0.5 mL wash buffer. Then, centrifugation process was repeated to obtain the pure mitochondrial fraction. After incubating the reaction mixture at 25°C for 3 min, 50 μL of the mitochondrial lysis suspension was added and immediately put into the spectrophotometer for detection. The absorbance at 420 nm and 470 nm was measured during 5 min to value the rate of FAO.
Measurement of biochemical parameters
Add 1 mL RIPA lysis buffer (Beyotime Biotechnology, P0013B) per 100 mg of heart tissue, homogenized on ice and centrifuged at 12 000 g for 20 min at 4°C. Transfer the supernatant to a clean tube as the total protein sample. Then reserve an aliquot of this supernatant for a protein assay by the BCA protein assay kit (Beyotime Biotechnology, P0012S). Plasma samples were collected via vacutainers containing sodium citrate and EDTA.
The cTn-I ELISA kit (E-EL-R1253c) was obtained from Biotech. The lactate dehydrogenase (LDH) assay kit (A020-2), creatine kinase MB isoenzyme (CK-MB) assay kit (E006-1-1), non-esterified free FA assay kit (A042-2-1), triglycerides (TG) assay kit (F001-1-1), total glutathione/oxidized glutathione (GSH/GSSG) assay kit (A061-1-2), and glutathione peroxidase (GSH-Px) assay kit (A005-1-2) were obtained from Nanjing Jiancheng Bioengineering Institute. The superoxide dismutase (SOD) assay kit (S0101S), glutathione (GSH) assay kit (S0053), and lipid peroxidation malondialdehyde (MDA) assay kit (S0131S) were obtained from Beyotime Biotechnology. The 8-isoprostane ELISA kit (516351) was obtained from Cayman Chemical. The pyruvate dehydrogenase kinase 4 (PDK4) ELISA kit (SEA958Hu) and peroxisome proliferator activated receptor alpha (PPARα) ELISA kit (SEA934Mu) were obtained from Cloud-Clone. All experiments were performed according to the manufacturer’s instructions, respectively.
Western blot analysis
Prestained protein marker (SMOBIO, PM2610) and total protein samples were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel, and then the proteins were transferred to polyvinylidene fluoride membrane overnight at 4°C. Membranes were blocked with 5% non-fat milk in Tris-buffered saline-Tween for 2 hours and were incubated with primary antibodies against CD36 (53KD, Sigma-Aldrich, SAB2100376, 1:500) and GAPDH (36KD, Abcam, ab8245, 1:1000) overnight at 4°C. After washing with phosphate-buffered saline Tween three times for 30 min, the membranes were then incubated with antirabbit fluorescent secondary antibody (1:15 000, CST, USA) for 2 hours at room temperature before scan in Odyssey imaging system (LI-COR, Nebraska, USA). The protein expression level was calculated from the optical density of the bands and normalized to GAPDH.
Statistics
Results are presented as mean±SD. The statistical test was performed by GraphPad Prism V.8.0 (San Diego). Comparison between the two groups was made with Student’s unpaired t-test. Comparison between multiple groups was performed with one-way analysis of variance (ANOVA) followed by Tukey’s test, or two-way ANOVA followed by Bonferroni’s test. A statistically significant difference was defined as p<0.05.
Results
Characteristics of diabetic rats
At the termination of the experiment, the daily water (figure 1A) and food intake (figure 1B), and the level of blood glucose (figure 1D) were significantly higher in the diabetes group than that in the non-diabetes group (all p<0.05). The diabetic rats had lower body weight (figure 1C) and plasma insulin level (figure 1E) than non-diabetic rats (both p<0.05). Similar to plasma free FA (figure 1F) and TG (figure 1G), the levels of myocardial free FA (figure 1H) and TG (figure 1I) were notably higher in diabetic rats than that in non-diabetic rats (both p<0.05). Besides, the expression of myocardial CD36 (figure 1J and L) and FAO rate (figure 1K) increased significantly in diabetic rats compared with non-diabetic rats (p<0.05).

Characteristics of diabetic rats (n=6). DM, diabetes mellitus; FAO, fatty acid oxidation; FFA, free fatty acid; TG, triglycerides. The Student’s unpaired t-test was used. *P<0.05.
Effects of diabetic conditions on the cardioprotection of IPostC
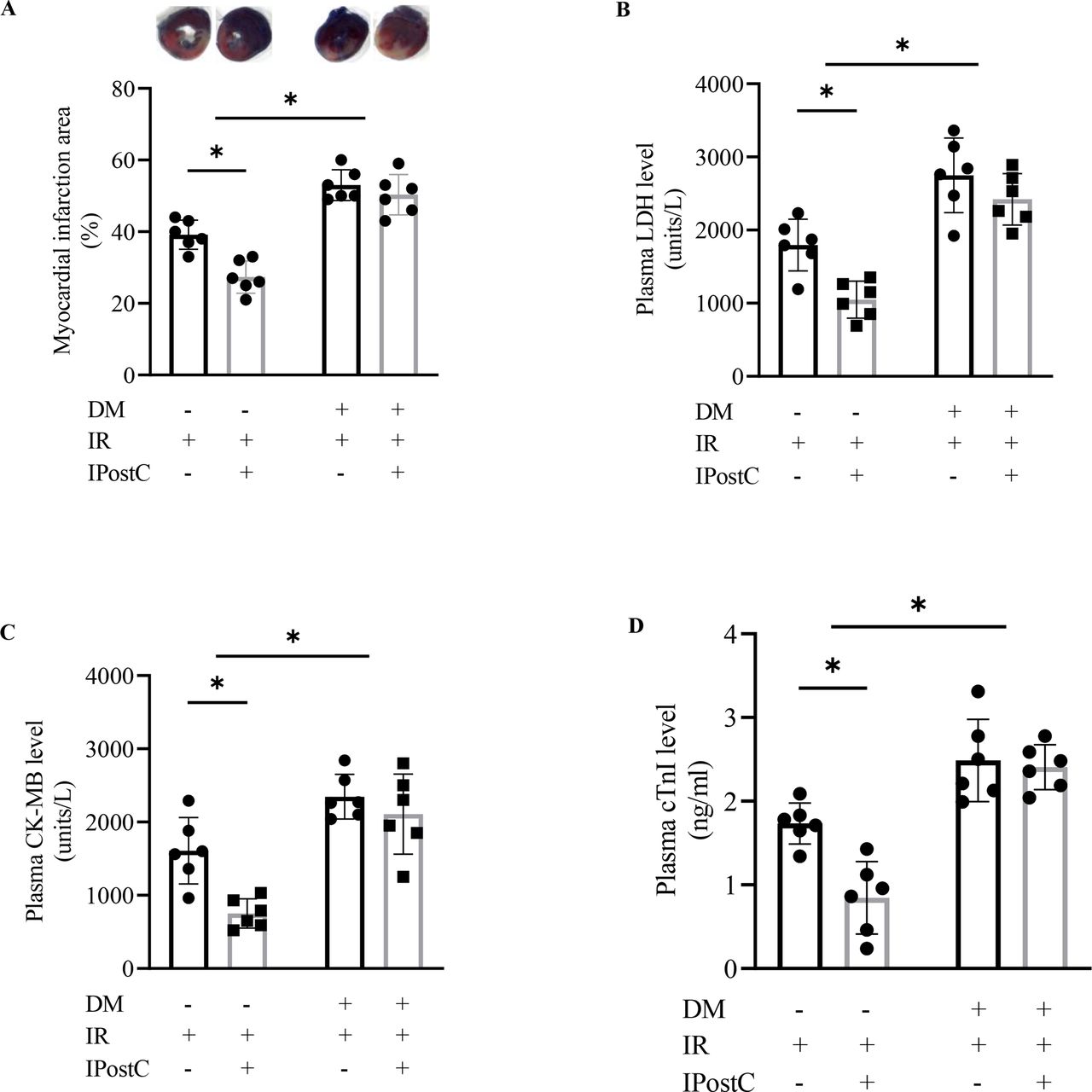
Compared with the non-diabetic rats, diabetic rats showed more serious myocardial injury after IR as evidenced by the significant increase in the ischemic size (figure 2A), the concentration of plasma LDH (figure 2B), CK-MB (figure 2C), and cTn-I (figure 2D) (all p<0.05). Importantly, IPostC significantly decreased the ischemic size and the levels of the concentration of plasma LDH, CK-MB, and cTn-I in non-diabetic rats (all p<0.05) but not in diabetic rats (all p>0.05).

Effects of diabetic conditions on the cardioprotection of IPostC (n=6). CK-MB, creatine kinase MB isoenzyme; DM, diabetes mellitus; IPostC, ischemic postconditioning; IR, ischemia reperfusion; LDH, lactate dehydrogenase. The two-way analysis of variance followed by Bonferroni’s test was used. *P<0.05.
Effects of SSO on the cardioprotection of IPostC in diabetic rats with myocardial IR
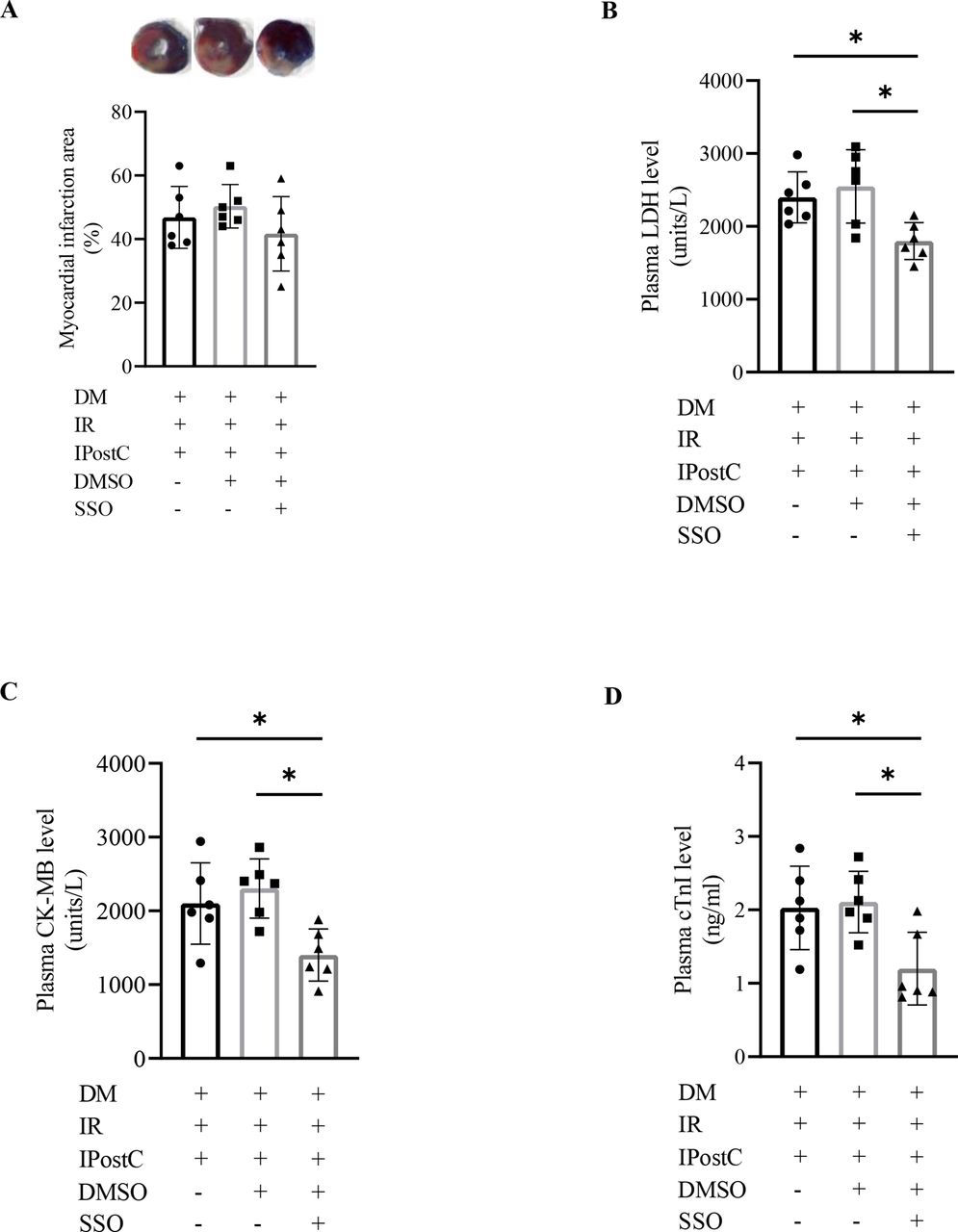
We wondered if SSO intervention could improve the cardioprotection of IPostC in diabetic rats suffering from myocardial IR injury. First, we detected the effects of DMSO on diabetic rats with myocardial IPostC. As shown in figure 3, no significant effects on the myocardial ischemic size and the concentration of plasma LDH, CK-MB, and cTn-I were observed in diabetic rats exposed to DMSO in the present setting (all p>0.05). Then, we treated diabetic rats with SSO under myocardial IR and IPostC conditions. The results did not display a significant difference in the myocardial ischemic size (p>0.05) (figure 3A), but in the level of plasma LDH (figure 3B), CK-MB (figure 3C), and cTn-I (figure 3D) between diabetic rats with or without SSO treatment during myocardial IR and IPostC (all p<0.05).

Effects of SSO on the cardioprotection of IPostC in diabetic rats (n=6). CK-MB, creatine kinase MB isoenzyme; DM, diabetes mellitus; DMSO, dimethylsulfoxide; IPostC, ischemic postconditioning; IR, ischemia reperfusion; LDH, lactate dehydrogenase; SSO, sulfo-N-succinimidyl oleate. The one-way analysis of variance followed by Tukey’s test was used. *P<0.05.
Effects of SSO on cardiac function in diabetic rats with myocardial IR and IPostC
As shown in figure 4, no significant change in cardiac function was observed between diabetic rats and non-diabetic rats. Myocardial IR resulted in significant increase in heart rate (figure 4A), but a significant decrease in dp/dtmax (figure 4E) and dp/dtmin (figure 4F) in diabetic rats. Although SSO treatment led to a significant decrease in E/A ratio (figure 4B) and a significant increase in dp/dtmin in diabetic rats with myocardial IR and IPostC (both p<0.05), there were no significant differences in the heart rate, ejection fraction (figure 4C), fractional shortening (figure 4D), and dp/dtmax among SSO-treated diabetic rats with IPostC and without (data not shown).

Effects of SSO on cardiac function in diabetic rats with myocardial IR and IPostC (n=6). E/A, the peak velocity of early (E) and late (A) diastolic filling. DM, diabetes mellitus; IPostC, ischemic postconditioning; IR, ischemia reperfusion; SSO, sulfo-N-succinimidyl oleate. The one-way analysis of variance followed by Tukey’s test was used. *P<0.05.
Effects of SSO on myocardial oxidative stress and FA metabolism in diabetic rats with myocardial IR and IPostC
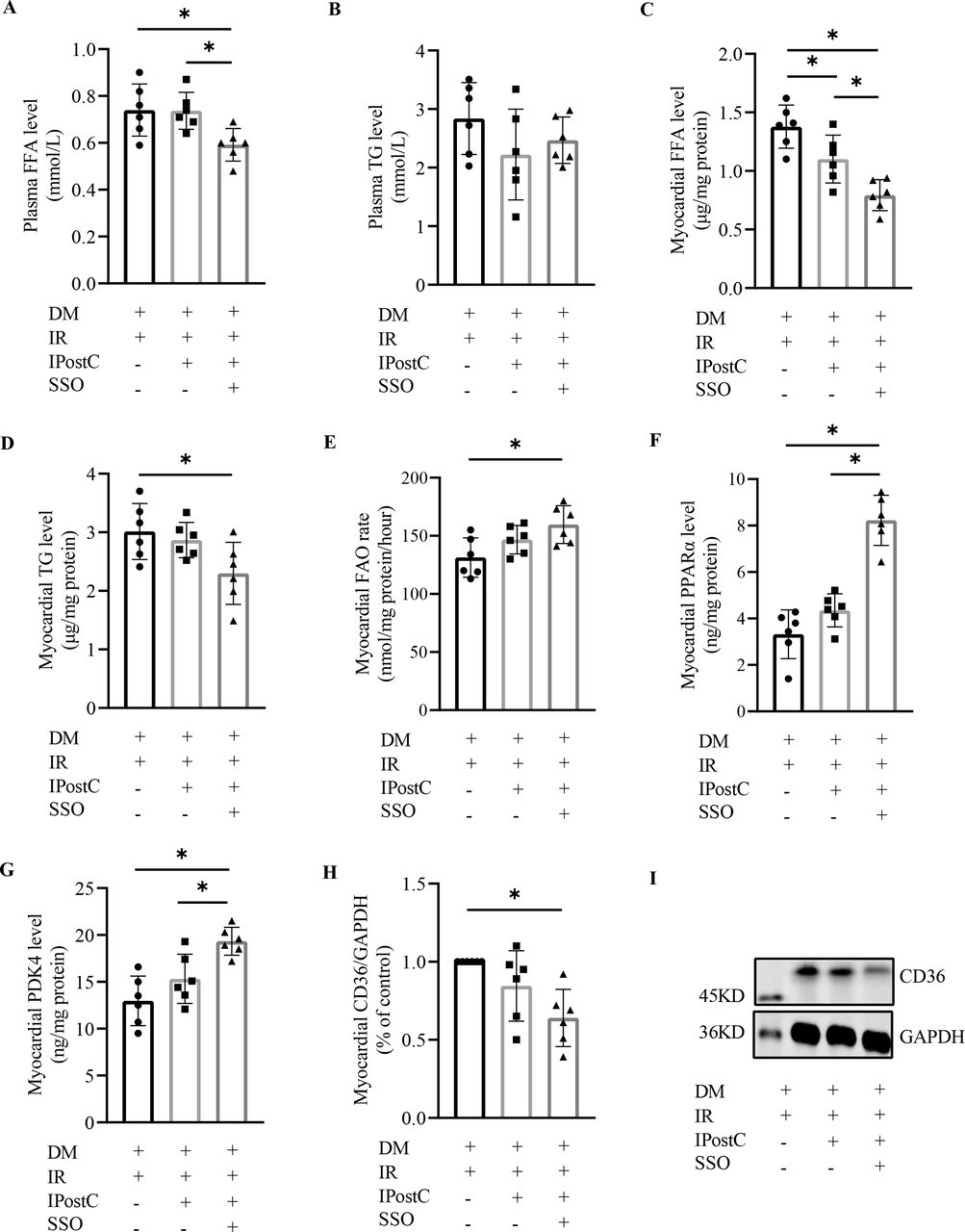
As shown in figure 5, there were no significant differences in the concentration of myocardial SOD (figure 5A), GSH (figure 5B), MDA (figure 5E), and 8-isoprostane (figure 5F) between diabetic rats with IPostC and without (all p>0.05), but not in myocardial GSH-Px level (figure 5C) and GSH/GSSG ratio (figure 5D). However, SSO intervention induced a significant decrease in myocardial MDA and 8-isoprostane content, and a further increase in myocardial GSH-Px level and GSH/GSSG ratio in diabetic rats with IPostC. Even though IPostC alone failed to ameliorate the FA metabolism effectively, IPostC combined with SSO intervention induced a significant difference in the concentration of plasma free FA (figure 6A), but not in plasma TG level (figure 6B). The level of myocardial free FA (figure 6C), TG (figure 6D), and CD36 expression (figure 6H and I) were significantly lower, but the level of myocardial FAO ratio (figure 6E), PPARα (figure 6F), and PDK4 (figure 6G) were significantly higher in diabetic myocardial IR rats with IPostC and SSO treatment than without.
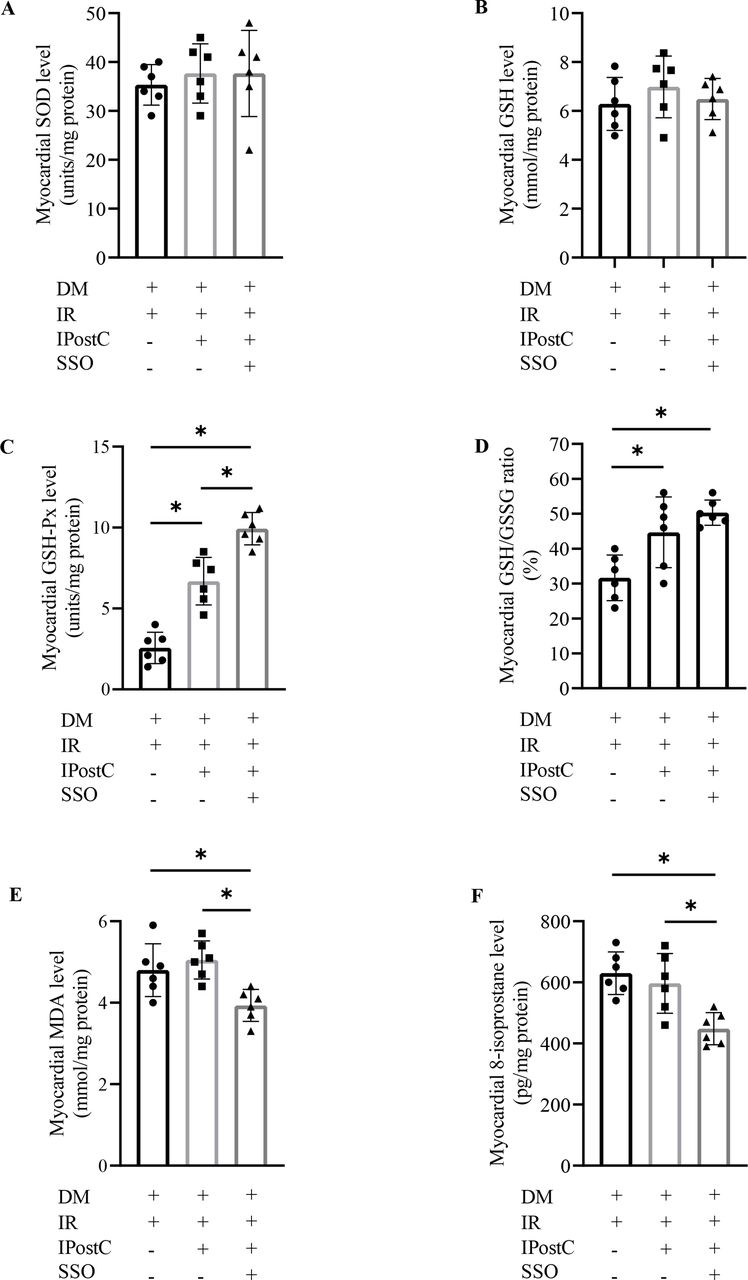
Effects of SSO on myocardial oxidative stress in diabetic rats with myocardial IR and IPostC (n=6). DM, diabetes mellitus; GSH, glutathione; GSH-Px, glutathione peroxidase; GSSG, oxidized glutathione; IPostC, ischemic postconditioning; IR, ischemia reperfusion; MDA, malondialdehyde; SOD, superoxide dismutase; SSO, sulfo-N-succinimidyl oleate. The one-way analysis of variance followed by Tukey’s test was used. *P<0.05.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of SSO on myocardial FA metabolism in diabetic rats with myocardial IR and IPostC (n=6). DM, diabetes mellitus; FAO, fatty acid oxidation; FFA, free fatty acid; IPostC, ischemic postconditioning; IR, ischemia reperfusion; PDK4, pyruvate dehydrogenase kinase 4; PPARα, peroxisome proliferator activated receptor alpha; SSO, sulfo-N-succinimidyl oleate; TG, triglycerides. The one-way analysis of variance followed by Tukey’s test was used. *P<0.0.
Discussion
It has been well demonstrated that myocardium requires increased ATP delivery by enhancing FAO when challenged with the increased workload, although glucose metabolism is also enhanced.14 In the diabetic myocardium, FA is the preferred substrate to support oxidative phosphorylation,15 due to compromised glucose metabolism increasing reliance on FAO.16 However, FA, as the oxidative substrate, generates more energy accompanied by more oxygen consumption than glucose. Consequently, the diabetic myocardium seems to be predisposed to anoxic damage. Consistent with other studies, our data showed that increased myocardial FAO rate was companied by decreased tolerance to IR injury and decreased susceptibility to IPostC in diabetic rats.
Clinical studies found that patients with diabetes exhibited an impaired ability of myocardial FAO even before the onset of contractile failure compared with patients without DM.17 CD36 polymorphism increased the risk of metabolic syndrome and acute myocardial infarction.18 Preclinical study found that CD36 was overexpressed in the myocardium of diabetic rats and high glucose-treated H9C2 cells. And, CD36 downregulation could inhibit cardiomyocyte apoptosis, which improved diabetic heart structure and function.4 Furthermore, cardiomyocyte-specific CD36 knockout mice showed significant decrease in myocardial FA uptake and TG accumulation compared with wild-type mice.19 Given that cardiac energy supply mainly depended on CD36-mediated FA metabolism due to CD36 accounts for over half of the total FA taken up in cardiomyocytes,3 20 maintaining appropriate FAO via regulating CD36 might represent a potential target in combating the negative effects of diabetes on the heart during stress conditions, such as IR injury. Interestingly, present data indicated that SSO combined with IPostC significantly increased myocardial FAO, decreased lipid accumulation, and CD36 expression in diabetic rats with myocardial IR.
The absence of CD36 has been shown to reduce brain injury size in the rats subjected to reperfusion after transient middle cerebral artery occlusion.21 22 Loss of CD36 also attenuated H2O2-induced lung microvascular endothelial cell injury in global CD36 knockout mice.23 These studies indirectly provided evidence for a CD36-dependent manner in cardioprotection against IR injury. Notably, global CD36 knockout was linked to a decrease in oxidized lipid product accumulation.24 CD36 could induce free radical production in the cerebral ischemia models, and antioxidant treatment attenuated CD36 overexpression-induced oxidative stress injury.25 Therefore, CD36 upregulation might be an important contributor to oxidative stress injury induced by diabetic myocardial IR.
Further study showed that cardiomyocyte-specific ablation of CD36 could improve postischemic functional recovery of the heart in non-diabetic mice.19 Myocardial IR enhanced FAO, accompanied by CD36 upregulation, which promoted ROS and 15-F2t-isoprostane formation in isolated non-diabetic rat hearts.26 Our study suggested that SSO-induced CD36 downregulation was accompanied by oxidative stress improvement and FAO enhancement in diabetic heart with IR and IPostC. Additionally, antioxidative treatment could recover sevoflurane postconditioning cardioprotection via inhibiting diabetic myocardial CD36 expression.12 The amelioration of oxidative stress via CD36 inhibition would be a valuable approach to alleviate diabetic myocardial IR injury.
A recent study has revealed that FA diffuse rapidly across biological membranes and do not require an active protein transporter for their transmembrane movement. SSO-induced FA uptake decrease might be due to the inhibition of oleate-triacylglycerol conversion in rat adipocytes.27 Previous study reported that CD36 downregulation by SSO treatment exerted cardioprotection against hypoxia/reoxygenation injury in isolated diabetic heart.11 The present study also found that SSO treatment partially attenuated myocardial IR injury in diabetic rats with IPostC via downregulating CD36 expression, improving FA metabolism and inhibiting oxidative stress. The beneficial effects of SSO in this context might be mediated by promoting FA oxidation to maintain appropriate energy supply and preventing excessive FA uptake and conversion to reduce lipotoxicity.28
Given the pharmacological activity of DMSO, this is a limitation of present study, although a relative lower dose was used. Previous studies have suggested that diabetic CD36-KO mice showed decreased tolerance to myocardial IR injury,29 further studies should get assessments on CD36-KO or conditional cardiomyocyte-KO mice.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Not applicable.
Ethics statements
Patient consent for publication
Ethics approval
All animal experiments were conducted in accordance with the guidelines of National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996), and approved by the Laboratory Animal Welfare & Ethics Committee (IACUC) of Renmin Hospital of Wuhan University.
References
Footnotes
YZ, HL, SS and LC are joint first authors.
YZ, HL, SS and LC contributed equally.
Contributors YZ and SS drafted the manuscript. HL and YZ analyzed and interpreted the work. HL and QM designed the work. SS, LC, and RC performed the experiments. QM and ZX reviewed it critically for important intellectual content. All authors contributed to the manuscript and approved the final version. MQT had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Funding This work was supported by the National Natural Science Foundation of China (81401574 and 81801085).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.