Article Text

Abstract
Introduction Mutations of CEL gene were first reported to cause a new type of maturity-onset diabetes of the young (MODY) denoted as MODY8 and then were also found in patients with type 1 (T1D) and type 2 diabetes (T2D). However, its genotype-phenotype relationship has not been fully determined and how carboxyl ester lipase (CEL) variants result in diabetes remains unclear. The aim of our study was to identify pathogenic variants of CEL in patients with diabetes and confirm their pathogenicity.
Research design and methods All five patients enrolled in our study were admitted to Shandong Provincial Hospital and diagnosed with diabetes in the past year. Whole-exome sequencing was performed to identify pathogenic variants in three patients with MODY-like diabetes, one newborn baby with T1D and one patient with atypical T2D, as well as their immediate family members. Then the consequences of the identified variants were predicted by bioinformatic analysis. Furthermore, pathogenic effects of two novel CEL variants were evaluated in HEK293 cells transfected with wild-type and mutant plasmids. Finally, we summarized all CEL gene variants recorded in Human Gene Mutation Database and analyzed the mutation distribution of CEL.
Results Five novel heterozygous variants were identified in CEL gene and they were predicted to be pathogenic by bioinformatic analysis. Moreover, in vitro studies indicated that the expression of CELR540C was remarkably increased, while p.G729_T739del variant did not significantly affect the expression of CEL. Both novel variants obviously abrogated the secretion of CEL. Furthermore, we summarized all reported CEL variants and found that 74.3% of missense mutations were located in exons 1, 3, 4, 10 and 11 and most missense variants clustered near catalytic triad, Arg-83 and Arg-443.
Conclusion Our study identified five novel CEL variants in patients with different subtypes of diabetes, expanding the gene mutation spectrum of CEL and confirmed the pathogenicity of several novel variants.
- Clinical Medicine
- Diabetes Mellitus, Type 1
- Diabetes Mellitus, Type 2
- Genetic Predisposition to Disease
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Clinical Medicine
- Diabetes Mellitus, Type 1
- Diabetes Mellitus, Type 2
- Genetic Predisposition to Disease
WHAT IS ALREADY KNOWN ON THIS TOPIC
Variants of the CEL gene can becausative for MODY and serve as a significant risk factor in chronicpancreatitis and pancreatic cancer, but its role in common forms of diabetes(i.e. type 1 and type 2) remains elusive. And there are only five families reportedin detail in the literature and these patients carried CEL frameshift variantswithin VNTR region. Moreover, very few CEL variants have been evaluated theirpathogenesis by functional studies.
WHAT THIS STUDY ADDS
Our present study first identified and reported five novel CEL variants in diabetic patients of different subtypes. And we exploited whole-exome sequencing, bioinformatic analysis as well as functional studies to characterize the pathogenic role of the novel CEL variants and summarized all previously reported mutations as well as their phenotypic information.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our results enrich the mutation spectrum of CEL gene to help elucidate CEL’s function and reveal the pathogenesis of CEL in diabetes.
Introduction
Monogenic diabetes refers to diabetes mellitus (DM) caused by a mutation in a single gene and accounts for approximately 1%–5% of diabetes.1 2 Maturity-onset diabetes of the young (MODY) is the most common type of monogenic diabetes characterized by autosomal dominant inheritance, early onset (typically <25 years old) and partial preservation of pancreatic β-cell function.3 Up to date, 14 subtypes of MODY have been identified, each caused by mutations in different genes.4 Although MODY represents 1%–5% of pediatric diabetes cases, it is frequently undiagnosed or misdiagnosed as type 1 (T1D) or type 2 diabetes (T2D) due to overlapping clinical characteristics. A correct diagnosis of MODY is of great clinical significance for patients because different subtypes also differ in clinical management.5 So genetic testing for MODY genes has the potential to provide more accurate diagnosis and more effective treatment for the patients.
MODY8 is caused by heterozygous mutations in the carboxyl ester lipase (CEL) gene, also known as bile salt-dependent lipase.6 Besides MODY8, CEL was also found to be associated with common forms of diabetes (ie, type 1 and type 2), chronic pancreatitis and pancreatic cancer.6–8 The enzyme CEL hydrolyzes dietary fat, cholesteryl esters and fat-soluble vitamins in the duodenum.9 10 CEL is mainly expressed in pancreatic acinar cells and lactating mammary glands.11 12 The human CEL gene resides on chromosome 9q34.3 and contains a variable number of tandem repeats (VNTR) region that encodes a mucin-like protein tail. There are two main domains of CEL: a globular N-terminal catalytic domain made up of 535 amino acid residues (excluding the signal peptide), followed by a C-terminal intrinsically disordered region including repeated 11-amino acid segments.13 14 The number of tandem repeats in humans ranges from 3 to 23, and most humans are homozygous for 16 repeats.15 16 The N-terminal region was responsible for catalytic activity including conserved catalytic triad Ser214-Asp340-His455.17 Similar to most mucinous proteins, the C-terminal region is mainly comprised of the unique 11-amino acid (KEAQMPAVIRF) repeats, which are enriched in the amino acids proline, glutamate, serine and threonine (PEST sequences).18 CEL’s C-terminus has been postulated to be critical for its secretion and activity.19 Until now, only 20 heterozygous CEL variants have been recorded in Human Gene Mutation Database (HGMD) to cause MODY8. Among them, there are only five families reported in detail in the literature and these patients carried CEL frameshift variants within VNTR region.6 20 21 However, few CEL variants have been evaluated their pathogenesis by functional studies. Since the genotype-phenotype correlation of CEL is still not clear, it will be helpful to better understand the genotype-phenotype relationship if more cases are found and studied.
Here we found five patients with diabetes with five new heterozygous variants of the CEL gene and confirmed the pathogenic roles of several variants through bioinformatic analysis and in vitro experiments. Our results enrich the mutation spectrum of CEL gene to help elucidate CEL’s function and reveal the pathogenesis of CEL in diabetes.
Material and methods
Study design
This study aimed to identify pathogenic variants in patients with MODY-like diabetes and evaluate the pathogenic role of the novel CEL variants. We first identified five novel CEL variant in five patients with diabetes through whole-exome sequencing (WES) and bioinformatic analysis suggesting that the novel variants were pathogenic. Then we assessed their impacts on CEL expression and its secretion in vitro. Finally, we summarized all previously reported mutations as well as their phenotypic information and analyzed the hotspots of CEL mutation.
Editorial policies and ethical considerations
The sampling and experimental procedures of the current study were performed by strictly adhering to the guidelines of the Helsinki Declaration 1964 and its latest amendments. Informed consent was obtained from all individual participants included in the study and written informed consent was received from participants prior to inclusion in the study.
Patients
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research. A detailed history about the onset and progression of diabetes as well as family history were obtained. Physical examination and laboratory detection were also performed to confirm the diagnosis. The diagnosis of MODY was considered in patients who met the following criteria: (1) atypical features of diabetes based on age <35 and insulin-independent within 2 years after diagnosis, (2) negative pancreatic antibodies, (3) a family history of diabetes consistent with autosomal dominant inheritance in at least two generations. Peripheral blood samples were obtained from the patients and their family members for genetic test.
DNA extraction and WES
Genomic DNA was isolated from peripheral blood leukocytes using the QIAamp DNA Mini Kit (Qiagen, Germany) following the manufacturer’s instructions. As for WES and subsequent Sanger sequencing for validation, we followed the methods of Wu et al.19 In addition, for patients 1 and 4, we performed real-time quantitative PCR for further validation. Genes and proteins were described according to the Human Genome Variation Society nomenclature guideline.
Bioinformatic analysis
To predict the potential pathogenic effects, we used three software tools, Mutation Taster (http://www.mutationtaster.org/), Poly Phen-2 (http://genetics.bwh.arvard.edu/pph2), PROVEAN (http:// provean.jcvi.org) to predict disease-causing effects of the mutation. Furthermore, AlphaFold Protein Structure Database (https://alphafold.com/) was used to search the structure of wild-type CEL and PyMOL software was used for visualizing the spatial structure and altered residues of CEL. The ClusPro server (https://cluspro.org) was also used to analyze protein-protein docking.
Plasmid construction
Wild-type and mutant (c.2187_2219delGGGTGACTCTGA GGCTGCCCCTGTGCCCCCCAC and c.1621C>T) human CEL plasmids (transcript ID: NM_001807.6) were synthesized using transient overexpression vector GV141 (GeneChem, China). The detailed protocol is available on request. The entire coding sequences of all the constructs were verified using sequencing.
Cell culture and transfection
We followed cell culture and transfection protocol from the literature.22 22 HEK293 cells (National Collection of Authenticated Cell Cultures, Shanghai, China) were grown in Dulbecco’s modified Eagle’s medium (Gibco BRL, Gaithersburg, Maryland, USA) containing 10% fetal bovine serum at 37°C in 5% CO2 and 95% air. Cells were seeded in 6-well plate prior to transfection and when they reached about 70% confluent, they were transfected with constructed wild-type and mutant CEL-GV141 overexpression vector or empty GV141 vector using Lipofectamine 3000 Transfection Kit (Invitrogen, USA). Transfection was performed for 12 hours with 2 µg plasmid per well.
Immunoblot analysis
We followed immunoblot protocol from the literature.22 22 HEK293 cells were rinsed with cold phosphate buffer solution (PBS), and whole cell lysates were lysed in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors. The cell media were collected, centrifuged and concentrated on Amicon Ultra-15 columns (MERCK) to get concentrated secreted proteins. Cell protein lysates (40 µg) and concentrated proteins (80 µg) in cell medium were separated using 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride membranes (Millipore). The membranes were blocked with Tris buffered saline tween(TBST) containing 5% slim milk for 1 hour at room temperature and then incubated with primary antibodies against Flag (1:1000, ProteinTech) and β-Actin (1:7500, ProteinTech) overnight at 4°C, after which they were incubated with secondary antibodies for 1 hour at room temperature. The membranes were incubated with horseradish peroxidase (HRP)-labeled secondary antibodies at room temperature for 1 hour, and Immobilon Western HRP Substrate Peroxide Solution (Millipore, USA) was used for membrane development.
Immunofluorescence assay
We followed immunofluorescence protocol from the literature.22 22 Forty-eight hours after transfection, cells were grown on glass coverslips and cell culture dishes were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and blocked for 1 hour in 2% bovine serum albumin (BSA). Immunostaining was accomplished with anti-Flag (1:300; ProteinTech) overnight at 4°C. Species-specific Alexa Fluor 488 secondary antibody (1:1000, Invitrogen) was used at room temperature for 1 hour. Nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole, blue). Protein localization was observed by fluorescence microscopy (Carl Zeiss, Germany).
Statistical analysis
Statistical analysis was performed by SPSS V.19.0 software package (SPSS). Continuous variables with normal distribution were given as mean±SD and compared by independent samples Student’s t-test. P value <0.05 was considered statistically significant.
Results
Clinical features
All of the participants involved in our study were of Han ethnicity. Three patients (patient 1/3/4) were clinically diagnosed with MODY, one diagnosed with T1D (patient 2) and T2D (patient 5). The clinical characteristics of all patients were summarized in online supplemental table 1. They were all males and aged from 1 to 48 years old. Patients 1, 3 and 4 were considered MODY because they had diabetes on age <35, negative antibodies and a family history of diabetes in three generations. Patient 2 was diagnosed with T1D because he had positive islet cell antibody and anti-glutamic acid decarboxylase antibody and a lack of insulin. Patient 5 was suspected of T2D because he demonstrated high level of C-peptide accompanied with metabolic syndrome. Moreover, except patient 2, all other patients have hyperlipidemia and patients 1, 3 and 4 have familial hyperlipidemia, especially hypertriglyceridemia, highly suggesting the presence of pancreatic exocrine dysfunction in these patients.
Supplemental material
Genetic analysis of CEL gene
To confirm the pathogenic gene of these five patients, they were subjected to WES and detected variants were further confirmed via Sanger sequencing or qPCR in them and their immediate family members including parents, siblings and offsprings. We found that patients 1 and 4 carried novel heterozygous Ex.8-11del and Ex.10-11del variants, respectively, and both had low levels of CEL transcripts compared with their unaffected family members carrying the full-length CEL (figure 1A and 1D). Patients 2 and 5 carried novel spontaneous variants c.830G>T (p.C277F) and c.1621C>T (p.R540C), respectively (figure 1B and 1E). Patient 3 shared a novel heterozygous c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC (p.G729_T739del) variant with his mother (figure 1C). All of them have not been reported in the HGMD, TOPMED, ExAC and 1000Genome databases, indicating that the mutations we have found were novel and rare. Interestingly, we found that all our patients with MODY carried deletion mutations, while both patients with T1D and T2D carried missense variants. Among five CEL variants, c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC (p.G729_T739del), Ex.8-11del and Ex.10-11del mainly affected the C-terminal intrinsically disordered region of CEL, leading to the truncation of C-terminal tail, while c.830G>T (p.C277F) and c.1621C>T (p.R540C) variants were located in the N-terminal catalytic domain of CEL and substituted original amino acid without changing the length of the protein (figure 2A).
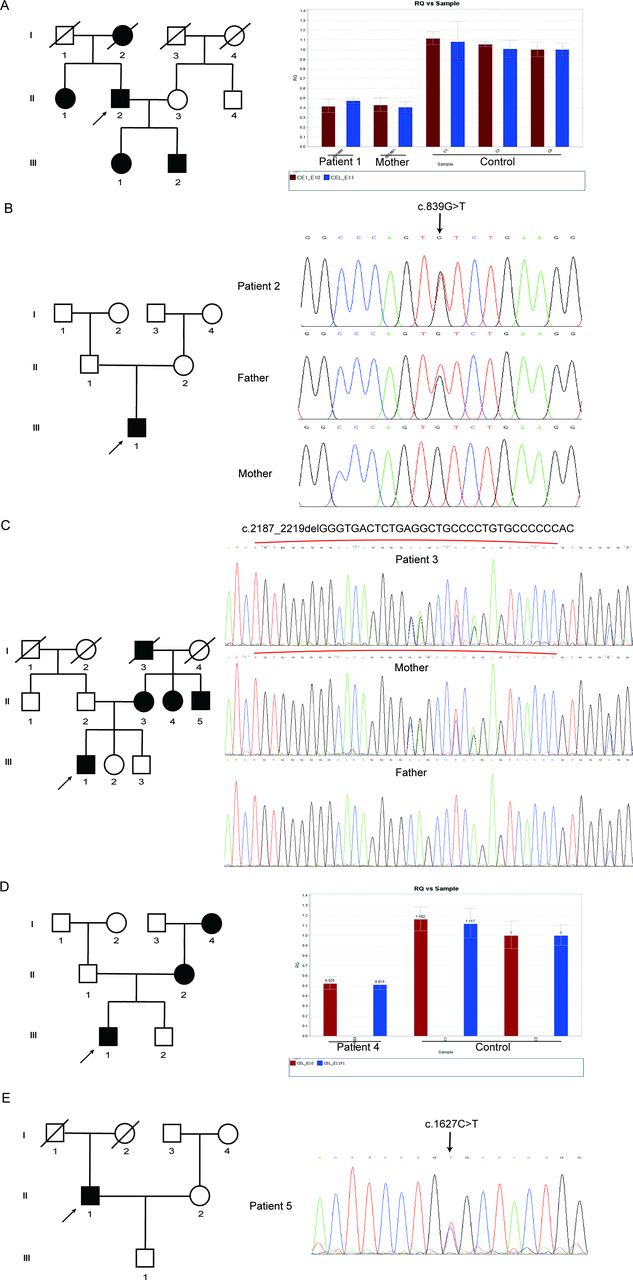
The pedigree of five patients with diabetes with novel carboxyl ester lipase (CEL) variants. (A) Pedigree of patient 1. Males and females are indicated by squares and circles. Affected individual is represented by filled symbols. The proband is represented by arrows. On the right is the original results of qPCR (Ex.10-11del) in the CEL gene. Red boxes represent the relative expression of the transcript of exon 10. Blue boxes represent the relative expression of the transcript of exon 11. (B) Pedigree of patient 2. On the right is the DNA sequence of the mutation site (c.830G>T/p.C277F) in the CEL gene. (C) Pedigree of patient 3. On the right is the DNA sequence of the mutation site (c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC/p.G729_T739del) in the CEL gene. Red brackets represented the mutated region. (D) Pedigree of patient 4. On the right is the original results of qPCR (Ex.8-11del) in the CEL gene. Red boxes represent the relative expression of the transcript of exon 10. Blue boxes represent the relative expression of the transcript of exon 11. (E) Pedigree of patient 5. On the right is the DNA sequence of the mutation site (c.1518C>T/p.R540C) in the CEL gene.

Schematic representation of CEL protein and bioinformatic analysis of two novel CEL missense variants. (A) Schematic structure of CEL protein. The amino acid numbers defining each domain are shown below. The newly identified variant is indicated in red. Purple represents N-glycosylation site; green represents catalytic sites. The drawing shows the most common human CEL with 16 tandem repeats in the C-terminal VNTR region. (B) Protein structure prediction of wild-type, CELC277F and CELR540C. Changed amino acids are marked in red. (C) Electrostatic potential of wild-type, CELC277F and CELR540C, negative and positive electrostatic potentials are shown as red and blue, respectively. (D) Predicted changes of the binding between CELR540C and GRP94. CEL and GRP94 was presented in blue and yellow, respectively. (E) Comparison of the sequences between the wild-type CEL (upper line) and the mutant protein (lower line). Dots indicate the same amino acid. The mutation caused the deletion of 729–739 amino acids and produced a truncated CEL. CEL, carboxyl ester lipase; VNTR, variable number of tandem repeats; WT, wild type.
Bioinformatic analysis
To clarify the pathogenic mechanism of these two novel CEL variants, we first performed bioinformatic analysis. c.830G>T (p.C277F) missense variant affected highly conserved amino acids in diverse species by multiple sequence alignment, highly suggesting it has disease-causing effects (figure 2B). c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC (p.G729_T739del), c.830G>T (p.C277F); Ex.8-11del; Ex.10-11del were strongly predicted to be pathogenic and deleterious using three online bioinformatic software—MutationTaster, PolyPhen-2, PROVEAN, while c.1621C>T (p.R540C) was predicted to be pathogenic by PolyPhen-2 but benign through PROVEAN and MutationTaster.
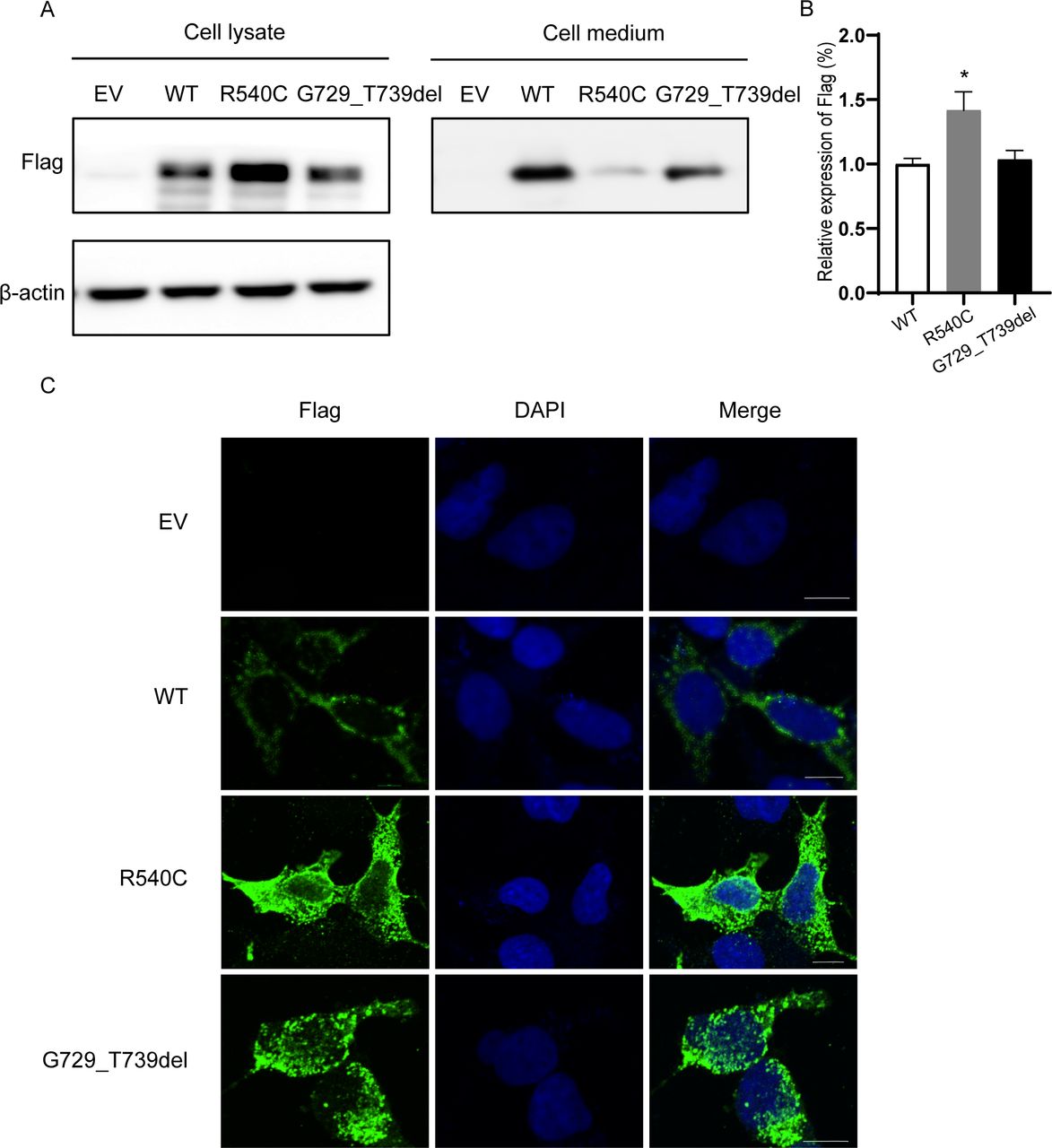
The protein model of wild-type CEL was downloaded from AlphaFold Protein Structure Database then the structures of mutant CEL were built and visualized by PyMOL viewer. As the structure of the C-terminal tail was unknown and was not able to be predicted based on the available databases, we could only predict the structure of CELC277F and CELR540C. We found that p.R540C variant did not significantly change the structure of CEL but the electrostatic potential at protein surface was remarkably altered, which might disturb the interactions of CEL with other macromolecules (figure 2B and C). Besides, we predicted the possible changes of the binding between CELR540C and glucose-regulated protein 94 (GRP94) which is a molecular chaperone interacting with CEL to form a CEL/GRP94 complex by ClusPro (figure 2D). p.G729_T739del variant leads to a deletion of 11 amino acids, resulting in a one-repeat shorter VNTR, which also has a reduced number of potential O-glycosylation sites (figure 2E).
Functional characterization of two novel CEL variants in vitro
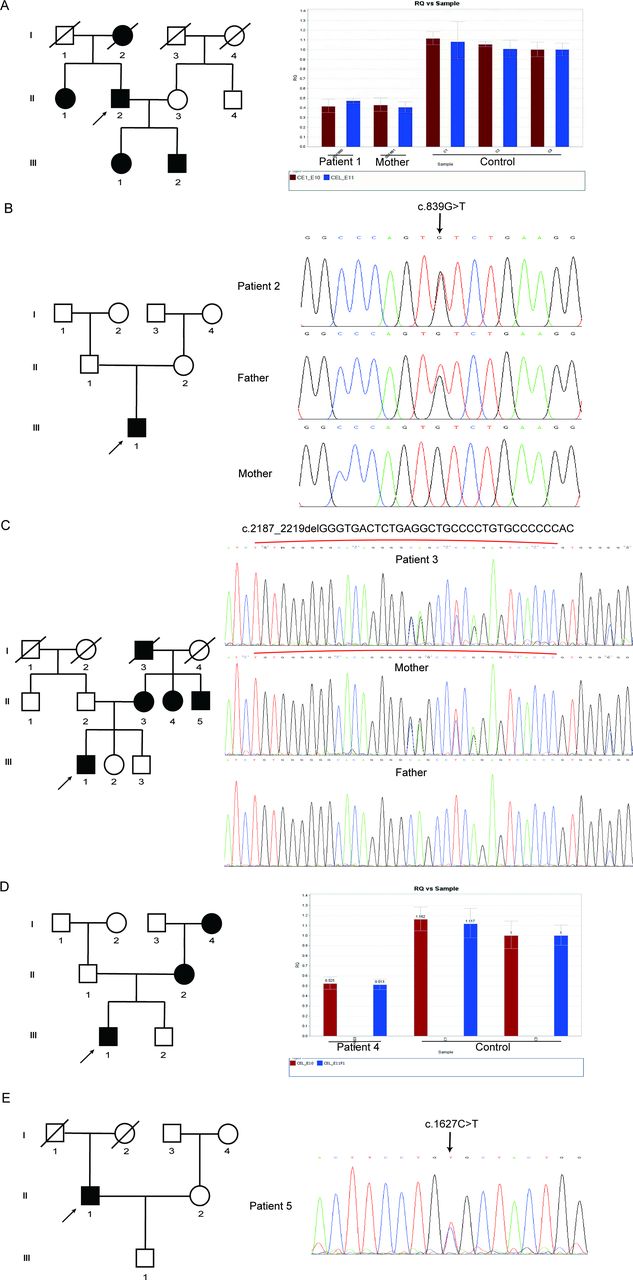
We chose two novel CEL variants to determine the functional effects. We transfected wild-type and mutant plasmids containing the wild-type CEL cDNA or CEL variants (NM_001807.6: p.G729_T739del; p.R540C) into HEK293 cells. First, we performed immunoblot analysis to assess the expression of CEL and the results showed that intracellular CEL of HEK293 cells transfected with plasmid carrying p.R540C variant was significantly increased while that of cells transfected with plasmid carrying p.G729_T739del variant was not obviously changed compared with wild-type group (figure 3A,B). As CEL functions as a secreted lipase, we also compared the CEL level in cell culture medium and we found that the protein levels of cells transfected with both mutant plasmids were remarkably decreased compared with wild-type group and the level of CELR540C was lower than CELG729_T739del (figure 3A). Then we performed immunofluorescence to evaluate the cellular localization of CEL and we observed a high tendency of intracellular aggregation of CEL in mutant groups (figure 3C). All of the above indicates that both novel CEL variants can significantly impair the secretion of CEL, leading to the intracellular retention of CEL.

Functional characterization of two novel CEL variants in HEK293 cells. (A) Expression pattern of wild-type and mutant CEL in HEK293 cells. HEK293 cells were transfected with CEL plasmids carrying WT-CEL or mutated CEL. The cell lysates and concentrated proteins in cell culture medium were fractioned on 10% SDS-PAGE and analyzed by immunoblotting with anti-Flag antibody. (B) Quantitative analysis of Flag expression levels. Shown is the mean percentage±SD of three biological replicates, * represented p<0.05 between WT and R540C groups (n=3). (C) Subcellular localization analysis of wild-type and CEL mutants in HEK293 cells. Forty-eight hours later, cells were fixed, permeabilized and immunostained with the anti-Flag antibody (green). Nuclei were visualized by DAPI. The slides were visualized on a fluorescence confocal microscopy (Leica, Germany). Scale bars represented 25 µm. Original magnification: x400. CEL, carboxyl ester lipase; SDS-PAGE, sodium dodecylsulfate-polyacrylamide gel electrophoresis; DAPI, 4′,6-diamidino-2-phenylindole; WT, wild type.
Summary of all CEL variants and mutation distribution in three-dimensional (3D) protein model of CEL
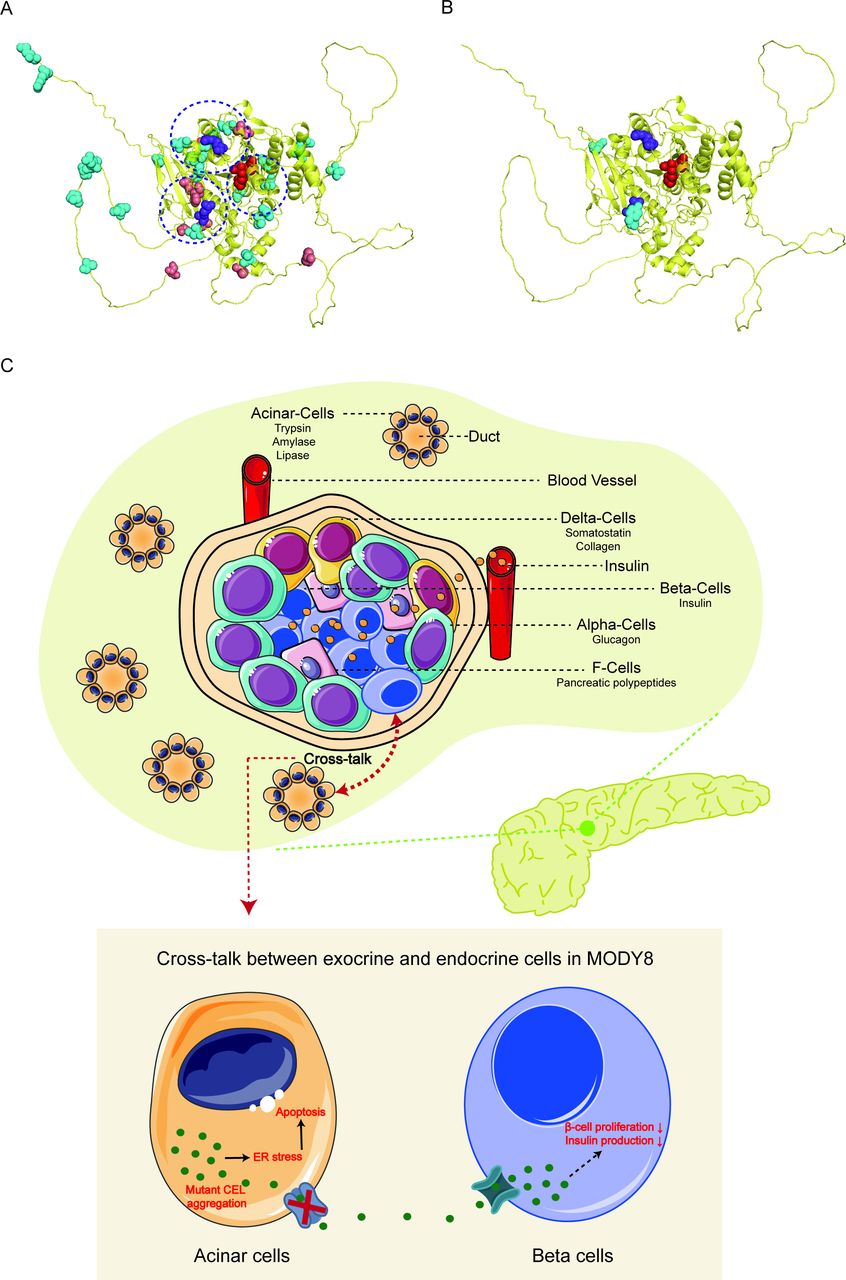
To visualize the distribution of the 38 missense and 2 nonsense CEL mutations identified in our study and previously reported in HGMD (online supplemental table 2), we used the 3D model of the CEL protein from AlphaFold Protein Structure Database (https://alphafold.com/). Interestingly, there were only two nonsense variants of CEL gene, located in the 95th and 113rd amino acid, respectively (figure 4B). Since nonsense variants tend to produce truncated protein which is more likely to be non-functional, they provide few insights into regions of the CEL protein which may have functional significance. It is worth noting that most missense variants were clustered near catalytic triad, arginine-83 and arginine-443 (circled by a dotted line) (figure 4A). Notably, arginine-83 and arginine-443 are important for modulating bile salt activation of CEL. Until now, 74.3% (29/39) of missense mutations were mainly located in exons 1, 3, 4, 10 and 11, indicating these are highly likely to be functional regions.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Distribution of missense/nonsense mutations in a 3D model of CEL. The 3D structure of CEL was from AlphaFold Protein Structure Database (https://alphafold.com/) (CEL, PDB ID: 6h19 A). (A) Missense mutations were relatively evenly distributed throughout the 3D CEL model. The dot-line circles represented clustered missense variants near arginine-83, arginine-443 and catalytic triad. Purple spheres represent Arg-83 and Arg-443. And red spheres represent catalytic triad (Ser214-Asp340-His455). Blue spheres represent point mutations related to MODY; orange spheres represent point mutations related to type 1 diabetes mellitus. (B) Only two nonsense mutations were located in the 95th and 113rd amino acid of CEL, respectively. (C) Possible mechanisms of MODY8 caused by CEL mutation. Cross-talk between acinar and β-cells potentially underlies the endocrine dysfunction in MODY8. In acinar cells, the expression of mutant CEL increased endoplasmic reticulum (ER) stress, activated the unfolded protein response and caused cell death by apoptosis. In addition, newly formed β-cells are surrounded by emerging acinar cells in the developing pancreas and this very close anatomical location probably enables long-term exposure of β-cells to mutant CEL protein synthesized by acinar cells in MODY8. Mutant CEL could impair the function and growth of β-cells, but the molecular mechanism is still unknown yet.
Discussion
It is known that MODY8 is an extremely rare disease caused by CEL gene mutation and inherited in an autosomal dominant manner. But little is known about the role of CEL in diabetes. In our study, we reported five patients with diabetes of different subtypes with five novel CEL pathogenic variants c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC/p.G729_T739del; c.830G>T/p.C277F; c.1621C>T/p.R540C; Ex.8-11del; Ex.10-11del. In silico analysis predicted both novel variants had pathogenic effects. And we also first explored the pathogenic mechanisms of two novel variants (c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC/p.G729_T739del; c.1621C>T/p.R540C) via in vitro experiments. Therefore, bioinformatics together with functional study provided strong evidence for the pathogenic roles of our new-found CEL variants.
The human CEL gene is ~10 kb in size and consists of 11 exons. There is a VNTR in the 11th exon which is made up of nearly identical 33 bp segments encoding the 11-amino acid repeats of the protein tail usually between 3 and 23 times.23 24 The most common type of human CEL has 16 repeated segments, thereby encoding a protein consisting of 745 amino acids with a predicted molecular mass of 79 kDa. Due to a varying number of C-terminal repeats in the general population and differences in post-translational modification, the observed molecular weight of the human CEL protein fluctuates significantly. CEL is N-glycosylated at a conserved Asn residue (Asn207) in the endoplasmic reticulum, then binding with molecular chaperone GRP94 for correct folding and secretion.25 Bioinformatic analysis showed that c.1621C>T (p.R540C) variant might abrogate the binding of CEL to GRP94, possibly resulting in misfolding and impeded secretion of CEL. The O-glycosylation sites are present in the C-terminal region of CEL which is comprised of 11-amino-acid region enriched in proline, glutamate, serine, and threonine (PEST sequence), which induces rapid protein degradation.18 O-glycosylation of the CEL repeats possibly masked PEST sequences, thus increasing the stability of CEL by preventing proteolysis.26 In addition, O-glycosylation of the CEL’s C-terminus has been demonstrated to be important for normal CEL secretion.27 As c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC (p.G729_T739del) variant was located in the C-terminus and produced a shorter, but still repetitive C-terminal, which also has a reduced number of potential O-glycosylation sites, which was highly likely to inhibit protein secretion and promote the degradation to cause disease. Once fully glycosylated at the trans-Golgi, CEL is phosphorylated by casein kinase II which promotes its release from intracellular membranes and drives the enzyme toward the secretion pathway.28 29 Different physicochemical properties of mutant CEL could disturb short-range and long-range interactions with other macromolecules. Ex.8-11del and Ex.10-11del variants lead to lower levels of CEL transcripts in patients; this might be attributed to significant degradation by nonsense mRNA-mediated decay.21
CEL is extremely polymorphic and has five categories of genetic variation: point mutation, VNTR repeats variation, small insertions or deletions mainly within the VNTR, splicing variants and copy number variants of the CEL locus. Up to date, there have been 64 CEL variants (39 missense/nonsense mutations; 2 splicing mutations; 10 small deletions; 7 small insertions; 3 gross deletions; 1 complex rearrangement and 2 VNTR repeat variants) recorded in the HGMD database. Among 39 point mutations, there are 37 missense variants and two nonsense variants. About 74.3% (29/39) of missense mutations were mainly located in exons 1, 3, 4, 10 and 11, and interestingly, exons 3 and 10 are critical regions for bile salt-binding and catalytic activity. Moreover, most missense variants clustered near catalytic triad, Arg-83 and Arg-443, which are key residues forming two parallel rows of positively charged residues near the active site domain of CEL necessary for CEL interaction with negatively charged micelles carrying the cholesteryl ester substrate.9
Variants of the CEL gene can be causative for MODY and serve as a significant risk factor in chronic pancreatitis and pancreatic cancer, but its role in common forms of diabetes (ie, type 1 and type 2) remains elusive.6–8 The genetic diagnosis of monogenic forms of diabetes (including MODY) are textbook cases of genomic medicine. In 2006, Ræder et al studied the single-bp insertions of the CEL VNTR (figure 3) in 182 adults with T1D or T2D. Within the diabetes cohort they found an association between single-bp insertions and low fecal elastase levels, first suggesting a role of CEL in the development of exocrine dysfunction in diabetes.6 Up to date, studies trying to bridge the gap between monogenic diabetes and common forms of diabetes (ie, T1D, T2D) have found a significant burden of pathogenic variants in genes related to MODY among patients with common T1D and T2D but the results are inconclusive.8 30 31 Until now, it has remained unclear how CEL variants cause diabetes. Previous studies found that a high tendency of both intracellular and extracellular aggregate formation of the mutants, indicating that MODY8 is a protein-misfolding disease.32–34 Notably, exocrine dysfunction is likely to be underdiagnosed in patients with diabetes. It has been shown that the prevalence of this complication is around 20%, at least when evaluated by fecal elastase test.35 Most previous fundamental studies only focused on the effects of the mutant CEL protein in acinar and non-endocrine cells as CEL mainly expressed in acinar cells. Recently, Kahraman et al provided compelling evidence for the mechanism by which a mutant gene expressed specifically in acinar cells promotes dysfunction and loss of β-cells to cause diabetes, indicating the pathogenic role of acinar cells in diabetes.36 But the specific molecular mechanism still requires further investigation.
To elucidate the pathogenic mechanism underlying the pathogenesis in our patients, we carried out a series of in vitro experiments. We found that the intracellular CELR540C was significantly increased compared with wild-type CEL. This could be attributed to either increased protein synthesis, decreased degradation or intracellular protein retention due to impeded secretion. Different from p.R540C variant, the expression of CELG729_T739del was not significantly changed but its level in the medium was remarkably reduced; this might be attributed to increased degradation of CEL according to previous studies. Moreover, we found that both CELR540C and CELG729_T739del mutants in the cell culture medium were remarkably decreased and the level of CELR540C was far less than that of CELG729_T739del, indicating that p.R540C variant might be more deleterious and damaging than p.G729_T739del variant. Consistently, we observed a high tendency of intracellular aggregation of mutant CEL. In vitro results were in accordance with the clinical characteristics of these two patients. In fact, the clinical phenotype of patient 5 was more severe than patient 3. Therefore, we proved intracellular retention of CEL due to impaired secretion of CEL was one pathogenic mechanism in our patients. A lack of integrated functional studies was a limitation in our study and we will investigate the biosynthesis, N/O-glycosylation, secretion and intracellular fate of the novel CEL variants in our further study. And future studies on the relationship between CEL and diabetes would be strengthened by the available results of pancreatic exocrine function in patients carrying CEL variants.
In summary, our study is the first to identify five novel CEL heterozygous variants (c.2187_2219delGGGTGACTCTGAGGCTGCCCCTGTGCCCCCCAC/p.G729_T739del; c.830G>T/p.C277F; c.1621C>T/p.R540C; Ex.8-11del; Ex.10-11del) in five Chinese patients with diabetes, which expands the mutation spectrum of CEL and promotes the understanding of its genotype-phenotype relationship. Moreover, the pathogenic consequences secondary to the variants help us to explain the underlying molecular mechanisms of CEL-related diabetes.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the Ethics Committee of Shandong Provincial Hospital (LCYJ: No. 2019-147). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors are sincerely grateful to all of the participants in this study. And the first author would like to heartfeltly thank her parents (Guiling Gan and Quanzhi Wu) for their maximum support in all aspects.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
HW and MS contributed equally.
Contributors CX acted as the guarantor to design and supervise the study as well as revise the manuscript. CL, QL, YS and TZ made the diagnosis and collected clinical data. YS and PS assisted the acquisition of clinical data. LF and RW assisted to perform genetic testing. WZ performed bioinformatic analysis. MS and XC performed in vitro experiments. HW performed data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Funding This study was supported by grants from the National Natural Science Foundation (No.81974124), together with special funds for Taishan Scholar Project (No. tsqn20161071).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.