Article Text

Abstract
Introduction Once-weekly subcutaneous semaglutide, a glucagon-like peptide-1 analog, is approved in the USA as an adjunct to diet and exercise for adults with inadequately controlled type 2 diabetes (T2D) to improve glycemic control and reduce the risk of major adverse cardiovascular events in people with T2D and established cardiovascular disease. The Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) phase III clinical trial program demonstrated the efficacy and safety of once-weekly subcutaneous semaglutide; however, determining its effectiveness in a real-world setting could support decision-making by clinicians, payers and policy makers in routine clinical practice.
Research design and methods SEmaglutide PRAgmatic (SEPRA) is an ongoing open-label, randomized, pragmatic clinical trial designed to compare the effects of once-weekly subcutaneous semaglutide versus standard of care in US health-insured adults with T2D and physician-determined inadequate glycemic control. The primary end point is the proportion of participants achieving glycated hemoglobin (HbA1c) <7.0% at year 1; other key outcomes include glycemic control, weight loss, healthcare utilization, and patient-reported outcomes. Individual-level data will be collected from routine clinical practice and health insurance claims. The last patient last visit is expected by June 2023.
Results Between July 2018 and March 2021, 1278 participants were enrolled from 138 study sites across the USA. At baseline, 54% were male with mean±SD age 57.4±11.1 years and body mass index 35.7±8.0 kg/m2. Mean diabetes duration was 7.4±6.0 years and mean HbA1c was 8.5±1.6%. At baseline, concomitant antidiabetes medications included metformin, sulfonylureas, sodium-glucose co-transporter-2 inhibitors, and dipeptidyl peptidase-4 inhibitors. The majority of participants had hypertension and dyslipidemia. The trial design was self-assessed using the PRagmatic Explanatory Continuum Indicator Summary-2 tool by the study steering group and was scored 4–5 in all domains suggesting a highly pragmatic study.
Conclusions SEPRA, a highly pragmatic ongoing study, will provide data on the effects of once-weekly subcutaneous semaglutide in a real-world setting when used during routine management of T2D.
Trial registration number NCT03596450.
Trial registration number
- glucagon-like peptide 1
- diabetes mellitus, type 2
Data availability statement
Data will be shared with bona fide researchers who submit a research proposal approved by the independent review board. Individual patient data will be shared in data sets in a de-identified and anonymized format. Data will be made available after research completion and approval of the product and product use in the EU and the USA. Information about data access request proposals can be found at novonordisk-trials.com.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The efficacy and safety of once-weekly subcutaneous semaglutide has been demonstrated by data from the phase III Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) randomized clinical trial program.
Once-weekly subcutaneous semaglutide was superior to both placebo and active comparators for reductions in glycated hemoglobin and body weight in a broad range of patient groups with inadequately controlled type 2 diabetes.
WHAT THIS STUDY ADDS
SEPRA will evaluate the effectiveness of once-weekly subcutaneous semaglutide versus other commercially available antidiabetes medications in a real-world setting when added to current oral antidiabetic therapy for individuals with inadequate glycemic control.
The data generated from the study will complement the findings of the SUSTAIN program.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Data from SEPRA may provide evidence to support decision-making by clinicians, payers, and policy makers in routine clinical practice.
The strategies used to mitigate the operational challenges encountered due to the pragmatic nature of the study may inform the design of future pragmatic clinical trials in diabetes and other chronic diseases.
Introduction
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) are a well-established treatment for people with type 2 diabetes (T2D).1 2 GLP-1RAs help individuals achieve glycemic control by increasing insulin and suppressing glucagon in a glucose-dependent manner, while also supporting weight loss by reducing appetite.3 4 Selected GLP-1RAs may also provide cardiovascular (CV) benefit in people with T2D.5 6
Semaglutide is a GLP-1RA approved in the USA for once-weekly subcutaneous use (as an adjunct to diet and exercise) in adults with inadequately controlled T2D to improve glycemic control, and to reduce the risk of major adverse CV events in adults with T2D and established CV disease.7–9 The efficacy and safety of once-weekly subcutaneous semaglutide was demonstrated by the phase III Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) randomized clinical trial program.8–14 In the SUSTAIN 1–7 clinical trials, once-weekly subcutaneous semaglutide was reported to be superior to both placebo and active comparators for reductions in glycated hemoglobin (HbA1c) and body weight in multiple patient groups with inadequately controlled T2D. A significant reduction in CV risk has also been reported for once-weekly subcutaneous semaglutide versus placebo (both as an adjunct to standard of care) in people with T2D and a high risk of CV events during SUSTAIN 6, a CV outcomes trial.9 A once-daily oral formulation of semaglutide (7 mg and 14 mg maintenance doses) is also available15 and has been evaluated in the Peptide InnOvatioN for Early diabEtes tReatment phase III clinical trial program.16–23 However, the present study focuses on once-weekly subcutaneous semaglutide only.
The current American Diabetes Association standard of care guidelines recommend adopting a patient-centered approach in the treatment of T2D.24–26 Pharmacotherapy should be initiated at diagnosis and tailored to the individual, accounting for comorbidities such as atherosclerotic CV disease, efficacy, impact on weight, cost and access, and individual preferences.24 First-line therapy generally includes metformin together with comprehensive lifestyle changes.24 In individuals with T2D with or at high risk of CV disease, GLP-1RAs or sodium-glucose co-transporter 2 (SGLT-2) inhibitors with proven CV benefit are recommended independently of background therapy (including metformin) and current or target HbA1c, to reduce the risk of CV events and mortality.24 GLP-1RAs may also be used as part of treatment intensification, if appropriate for the clinical needs of the individual (eg, where it is beneficial to provide additional HbA1c control, to avoid hypoglycemia, or to minimize weight gain or promote weight loss).24
To inform decision-making by clinicians, payers, and policy makers in routine clinical practice, the ongoing SEmaglutide PRAgmatic (SEPRA) clinical trial is a comparative effectiveness study of treatment intensification of current antidiabetic therapy with either once-weekly subcutaneous semaglutide or any other medication indicated for diabetes treatment at the discretion of the treating provider, hereafter termed ‘standard of care’. Eligible participants were diagnosed with T2D and treated with two oral antidiabetes medications, but required additional medication as determined by the provider in a variety of practice settings in the USA. Pragmatic clinical trials are used to generate evidence on the effectiveness of an intervention in routine clinical practice, while explanatory clinical trials are conducted in an idealized setting to provide the optimum scenario for a treatment to show a beneficial effect.27
Here, we describe the design of the SEPRA trial and present the baseline data collected from participants enrolled, including participant demographics and clinical characteristics, as well as comorbidities and concomitant oral antidiabetes medications. We also present the findings of the PRagmatic Explanatory Continuum Indicator Summary-2 (PRECIS-2) analysis and discuss how we overcame recruitment challenges encountered due to the pragmatic nature of the study.
Methods
Trial design
SEPRA (NCT03596450) is an ongoing, randomized, open-label, phase IV pragmatic clinical trial that was designed to compare the effects of once-weekly subcutaneous semaglutide versus standard of care when added to up to two oral antidiabetes medications, as treatment intensification among adults with T2D during routine clinical practice in the USA (figure 1).

Study design of the SEmaglutide randomized PRAgmatic trial.
Participants were recruited from 138 physician sites across the USA between July 2018 and March 2021. The last patient last visit is expected by June 2023.
Measurement of pragmatic elements
The pragmatism of the SEPRA study design, prior to any protocol amendments, was qualitatively assessed using the PRECIS-2 tool by the study steering group at a workshop held in November 2018. The study steering group included 11 members from HealthCore, Novo Nordisk and independent expert advisors who were involved in protocol development. The PRECIS-2 tool has nine domains including eligibility, recruitment, setting, organization, flexibility (delivery), flexibility (adherence), follow-up, primary outcome, and primary analysis.27
Each member of the steering group independently assessed the study design prior to the workshop using the PRECIS-2 criteria and the independent assessments were subsequently collated and shared with the group for discussion. Participants were given the opportunity to provide their rationale and a consensus rating was reached for the nine domains during the discussion. The methods are described below as per the PRECIS-2 domains.
Eligibility domain
In the initial protocol, the inclusion and exclusion criteria were minimally restrictive to allow recruitment of a broad population of participants with a focus on the need for T2D treatment intensification and no prior use of semaglutide, as shown in box 1. During the recruitment period, enrollment rates were lower than projected, and the eligibility criteria were subsequently amended to expand the population recruited from each site (box 1; figure 2). The first key amendment in March 2019 allowed for enrollment of participants on up to two oral antidiabetes medications rather than metformin alone and the second key amendment in August 2019 allowed the enrollment of participants with any health plan with pharmacy benefits. The eligibility criteria were further amended in December 2019 to specify the exclusion of participants receiving oral semaglutide. If patients are not started on study medication this is considered a protocol violation, but the participant will be included in the analysis dataset.
Study inclusion and exclusion criteria
Original eligibility criteria (March 2018)
Adult participants (≥18 years) with type 2 diabetes (T2D) treated with metformin monotherapy.
Requirement for further treatment intensification for glycemic control with an additional antidiabetes medication (treating study physician determined) as per the Food and Drug Administration-approved subcutaneous semaglutide label.7
Current member of an Anthem-affiliated commercial health plan with pharmacy benefits.
Recorded glycated hemoglobin value within the last 90 days prior to randomization.
No previous randomization in the study.
No treatment with any medication indicated for diabetes other than metformin in the 30 days before eligibility assessment.
No contraindications to semaglutide (as according to the Food and Drug Administration-approved label).
For women, not being pregnant, breast feeding or intending to become pregnant.
No participation in another clinical trial.
Amended eligibility criteria (March 2019)
Adult participants (≥18 years) with T2D treated with one or two oral antidiabetes medications.
Amended eligibility criteria (August 2019)
Current member of any health plan with pharmacy benefits.
Amended eligibility criteria (December 2019)
Adult participants (≥18 years) with T2D treated with one or two oral antidiabetes medications, excluding oral semaglutide.

Projected and actual recruitment rate. Amendment 1 included enrollment of participants on ≤2 oral antidiabetes medications; amendment 2 included enrollment of participants with any health plan with pharmacy benefits. OADs, oral antidiabetes medications.
Setting and recruitment domain
Potential physician sites, including both primary care practitioners and endocrinologists, were selected by querying the HealthCore Integrated Research Database (HIRD) to identify eligible individuals (ie, those with Anthem-affiliated commercial health plans with pharmacy benefits) and subsequently mapping back to healthcare providers. The HIRD is a large administrative healthcare database containing longitudinally integrated medical and pharmacy claims data from commercially insured individuals across the USA (from January 1, 2006 to present). Following recruitment challenges, the protocol was updated in August 2019 to allow participation of sites with prior research experience with semaglutide.
Eligible individuals were invited to participate in the study when they presented to their physician during routine clinical care and through proactive identification from within the study site patient population. The assessment that an individual had inadequate glycemic control on up to two oral antidiabetes medications was made by the treating study physician prior to, and independently of, study enrollment and prior to signing informed consent. On determining a need for treatment intensification, the physician assessed suitability according to the current eligibility criteria and the approved label for once-weekly subcutaneous semaglutide.
Organization (randomization and trial regimen) domain
This is an open-label study in which randomization was included to reduce selection bias and ensure comparable patient populations in the two treatment groups. There is a 4-week screening period during which time the treating physician can confirm the need for antidiabetic therapy intensification and ensure the participant meets eligibility criteria. Participants are then randomized in a 1:1 ratio with permuted blocks of size four using centralized allocation via the study electronic data capture system to be prescribed either once-weekly subcutaneous semaglutide or standard of care, both as add-on to up to two oral antidiabetes medications, on enrollment to the study (online supplemental figure 1). Standard of care is defined as a single mixed comparator arm that follows routine clinical practice most closely (as patient and doctor preferences/prescribing determined the mix of treatments in this arm), and thus renders higher generalizability to settings where a similar mix of usual care treatments is used. Furthermore, in a trial with a long duration (in this case 2 years), standard of care may change during the conduct of the trial, for example, due to changes in reimbursement or if a new medication becomes available on the market. In this situation, changes in usual care in newly recruited patients, or switches to a new usual care regimen in enrolled patients, may be appropriate to continuously reflect routine clinical practice. Otherwise, generalizability may decrease.
Supplemental material
Standard of care includes addition of any commercially available oral or injectable antidiabetes medications, other than semaglutide, prescribed at the discretion of the physician for antidiabetic treatment intensification following randomization. Commercially available GLP-1RAs, except semaglutide, could be prescribed. The study drug in the standard of care group was defined as the drug class of the first antidiabetes oral or injectable medication prescribed for treatment intensification following randomization. In the event that a fixed-dose combination product was prescribed, the treating study physician (ie, the participant’s own physician enrolled in the study) chose one to be the study drug. Participants were not permitted to switch to semaglutide at any point during the study period.
Participants are prescribed once-weekly subcutaneous semaglutide or another standard of care medication based on the randomization allocation by the treating physician via routine prescribing methods at the time of the randomization visit. Postrandomization diabetes care is managed by their own treating physician, who adjusts treatment according to their own clinical judgment.
Flexibility (delivery and adherence) domains
Each treating study physician is responsible for making treatment decisions according to their clinical judgment and knowledge of their patient. Participants randomized to the once-weekly subcutaneous semaglutide group are being prescribed subcutaneous semaglutide in a prefilled pen injector, with semaglutide initiated according to approved labeling. Add-on, discontinuation, or dose modification of oral antidiabetes medications, including subcutaneous semaglutide, during the study are at the discretion of the treating study physician.
In both treatment groups, prescriptions for randomized study drug are being handled and dispensed by a pharmacy of the participant’s choice per routine care, in line with their preference and health plan benefits. All participants are responsible for paying an equalized (ie, the same amount for once-weekly subcutaneous semaglutide arm and alternative antidiabetes medications in the standard of care arm) out-of-pocket maximum cost of US$20/month. This is to minimize the impact of any differential out-of-pocket costs between the treatment groups influenced by variations in individual health plan design and benefits. The participants’ out-of-pocket cost will be up to the specified maximum for the randomized study drug and ancillary needles (if required to administer the study drug), and the sponsor will reimburse additional costs above this maximum related to randomized study drug. Payment is processed at the pharmacy.
Follow-up domain
Treating study physicians or site personnel are collecting patient characteristics and study data at each visit, either directly from the patient or from the patient’s medical records, and entering them into the electronic case report form.
Participants will be followed up for 2 years after randomization, regardless of changes in antidiabetes medication over the course of the study, unless informed consent is withdrawn. Medical and pharmacy claims data will be extracted from the HIRD and other administrative claims databases for the 2-year study period, as well as up to 12 months prior to randomization, where available. These data are not anticipated to be available for all patients.
Outcome domain (study end points and assessments)
The primary end point is the proportion of participants who achieve HbA1c <7.0% (53 mmol/mol) at year 1. Confirmatory secondary end points and other supportive end points, including patient-reported outcomes (PROs) and clinician-reported outcomes, are listed in online supplemental table 1 and appendix 1.
Diabetes treatment satisfaction, generic health-related quality of life, work productivity, and patient and clinician global assessments will be assessed throughout the study. The tools employed include the Diabetes Treatment Satisfaction Questionnaire; Short Form 12-Item version 2 (V.2) Health Survey; Work Productivity and Activity Impairment: General Health questionnaire; the Patient Global Impression of Disease Severity and Patient Global Impression of Change scales; and the Clinician Global Impression of Disease Severity and Clinician Global Impression of Change scales, described in online supplemental appendix 1.28–33 Paper-based PROs will be completed by each patient, either in person or mailed to the study site, and reviewed for completeness by site study personnel before responses are entered into the electronic case report form.
Serious adverse events, adverse events leading to study drug discontinuation, and pregnancies will be collected and coded using the Medical Dictionary for Regulatory Activities and descriptively summarized by System Organ Class and Preferred Term.
Organization and intervention domain (data collection)
Primary data are collected prospectively at study visits and include demographic and clinical data, participant-completed PRO data, and clinician-reported global assessments. Secondary data are collected from administrative claims data from health plans, where available.
Dedicated study visits are taking place at randomization, year 1, and year 2. Any other visits during the study are routine clinical visits, including office visits and other participant contacts. Clinical data are also collected at these routine visits (assessments are described in online supplemental table 2).
Analysis domain (statistical analysis)
Two different scientific questions related to the efficacy objectives will be addressed through the definition of two estimands: ‘intention-to-treat (ITT)’ and ‘if all participants had adhered’. The primary estimand for all end points is the ITT estimand, which evaluates the effectiveness of randomized treatment intervention, irrespective of adherence or changes to other antidiabetes medications. The secondary estimand for all end points, except for the adherence and persistence to treatment objective, is the ‘if all participants had adhered’ estimand. This estimand evaluates the effect of randomized treatment intervention for all randomized participants if all participants had adhered to randomized treatment, regardless of changes to other antidiabetes medication.
At study initiation, the planned enrollment was 2250 participants to provide 90% power to jointly confirm superiority of the primary end point and the three confirmatory secondary end points. The target sample size was subsequently revised to 1387 participants, which aims to provide 90% power to confirm superiority of the primary end point and 85% power to also confirm superiority of the first confirmatory secondary end point (based on an analysis of the primary estimand for each of the end points).
Demographic and baseline characteristics were summarized using descriptive statistics.
Results
Enrollment
Overall, 138 physician sites were enrolled to the study from across the USA (online supplemental table 3). Of these sites, the majority are primary care clinics (72.5%) and the others are endocrinology care clinics (27.5%), and less than one out of four sites have prior experience with semaglutide research.
Baseline profile of the total study population
From July 2018 through March 2021 (a recruitment period of 33 months), a total of 1312 participants were screened, of whom 1278 were randomized, following which the site initiated the enrollment process.
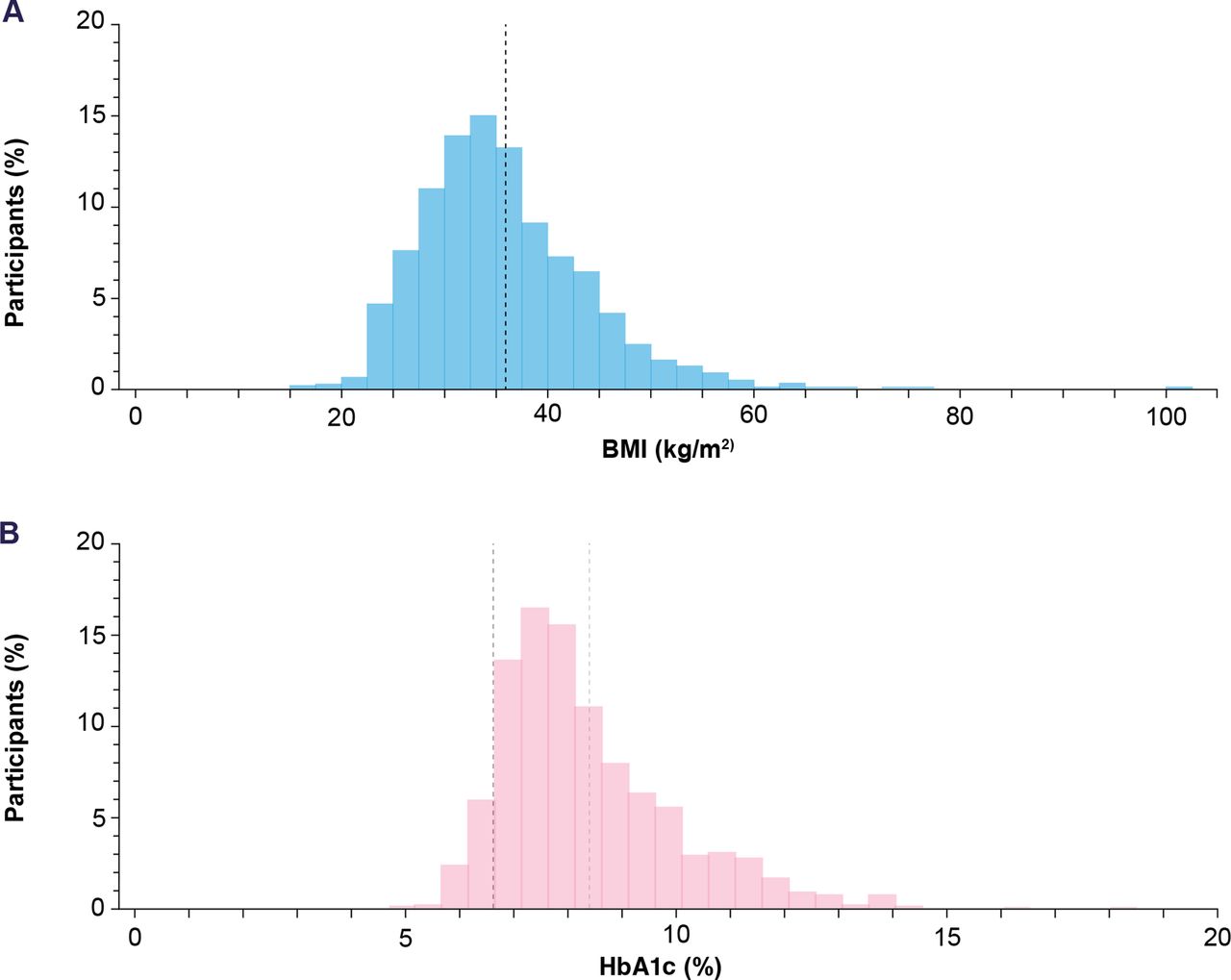
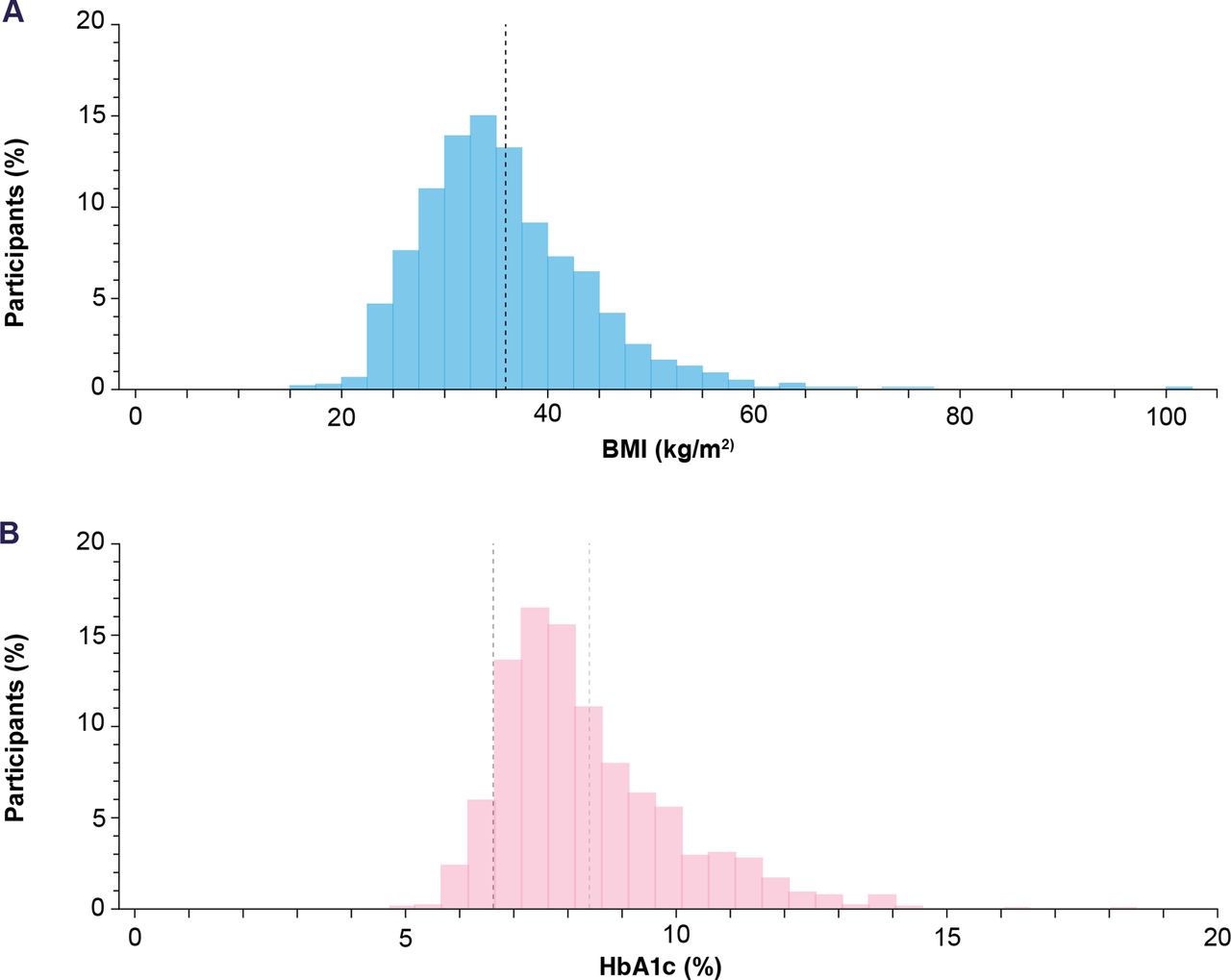
Participants with high variability across a broad range of demographic and clinical characteristics were enrolled. Of the 1278 participants enrolled, just over half (54.2%) were male and the majority (78.6%) were white. The mean (±SD) age was 57.4±11.1 years and 25.8% (330/1278) were aged 65 years or over. The overall mean (±SD) duration of T2D was 7.4±6.0 years (table 1). The overall mean (±SD) body mass index (BMI) was 35.7±8.0 kg/m2, which was broadly distributed across a range from 17.0 to 100.6 kg/m2 (figure 3A). At baseline, the mean (±SD) HbA1c was 8.5±1.6% (69.2 mmol/mol). Baseline HbA1c values ranged from 4.9% to 18.5%, showing a wide range of glycemic control with substantial representation of elevated HbA1c levels (figure 3B).
{kind=link}
{kind=link}
{kind=link}
Histogram of mean BMI (range: 17.0–100.6 kg/m2) (A) and HbA1c (range: 4.9%–18.5%) (B) at baseline. (A) The black dashed line indicates the mean (SD) BMI (35.7±8.0 kg/m2). (B) The black dashed line indicates the mean (SD) individualized HbA1c target (6.7%±0.5), and the light grey line indicates the mean HbA1c (8.5%±1.6). BMI, body mass index; HbA1c, glycated hemoglobin.
Baseline demographic and clinical characteristics of participants
Concomitant medications and comorbidities
Concomitant antidiabetes medications at baseline were metformin (88.7%), sulfonylureas (20.8%), SGLT-2 inhibitors (15.2%), dipeptidyl peptidase-4 inhibitors (10.9%), and thiazolidinediones (2.7%). The majority (85%) of participants were receiving concomitant CV medications at baseline (table 1). The most frequently used (reported in >10% of participants) were statins (55.5%), ACE inhibitors (33.2%), angiotensin receptor blockers (22.6%), beta-blockers (20.4%), aspirin (18.8%), calcium channel blockers (16.4%), and thiazide diuretics (13.2%). The majority of participants (92.6%) had a comorbid condition, the most common of which were hypertension (77.2%) and dyslipidemia (71.3%) (table 1).
PRECIS-2 assessment
The trial design was retrospectively assessed using the PRECIS-2 tool27 by the study steering group in December 2018. The allocated scores were plotted in a PRECIS-2 wheel (which is similar to a radar or spider chart) showing that the study was scored 4–5 in all nine domains (1=very explanatory and 5=very pragmatic) (online supplemental figure 1; online supplemental table 4).
Eligibility criteria domain
Eligibility criteria, defined as how strict (explanatory) or open (pragmatic) the eligibility criteria for the trial are, was rated as 4. This was based on the rationale that study participants were enrolled during routine clinical care with limited exclusion criteria, following the decision of treating study physicians to intensify antidiabetes treatment based on their clinical judgment to achieve individualized glycemic targets.
Recruitment domain
Recruitment was rated as 5. Recruitment efforts were limited to reminder calls from HealthCore to the study sites and efforts were made to avoid interrupting the usual flow of standard care as individuals were recruited during their routine care and interactions with the site.
Setting domain
Setting, defined as where the trial is being conducted, was rated as 5. The study was performed within settings in which the study participants received their routine clinical care.
Organization domain
Organization was defined by how much expertise and additional resources the physician requires to execute the trial, including both infrastructure and the knowledge needed to deliver the intervention. A consensus was reached on a score of 4. Treating study physicians did not need to have large research infrastructure to complete the trial as there were minimal study visits, targeted site data collection, and no requirements for study medication storage/dispensing. The study interventions were all US Food and Drug Administration-approved antidiabetes medications. It was also noted that compliance and persistence could be influenced by participants being aware of participating in a pragmatic clinical trial versus what might be observed in real-world practice.
Flexibility domains
Flexibility (delivery), defined as how the intervention should be delivered, was scored 5. Flexibility (adherence), defined as what measures are in place to ensure participants adhere to the intervention, was scored 5. Treatments are prescribed via the treating study physician in line with approved indications. Prescribed treatments are dispensed by a pharmacy of the participant’s choice reflecting routine clinical care. Participants chose their own pharmacy per usual care to receive their medications, were responsible for paying an equalized out-of-pocket cost to mimic the typical prescription fill process, and no study-specific medication adherence methods were employed.
Follow-up domain
Follow-up, defined by how closely participants are followed up via visits and assessments, was scored 4, based on the rationale that there are three protocol-mandated visits that would not usually occur during routine clinical care. Furthermore, questionnaires assessing quality of life and other PROs are also not typically part of routine clinical practice.
Primary outcome domain
The primary outcome domain, defined as how relevant the end points and results are to trial participants, was scored 4. The steering group reported that composite end points are not considered to be highly pragmatic. The end point of the proportion of participants achieving HbA1c <7.0% was considered a payer-centric end point and not directly relevant to participants, but it has been reported that individuals with diabetes do regard HbA1c as an important metric.11 In real-world practice and supported by treatment guidelines,24 flexibility is often applied to these cut-offs. The inclusion of secondary end points such as achievement of individualized HbA1c targets determined by the treating study physician before randomization and change from baseline in HbA1c was deemed highly pragmatic. Measuring use of healthcare resource utilization is both relevant and pragmatic.
Primary analysis domain
Primary analysis, defined by what data are included in the analyses, was rated as 5 as the ITT population will be used for at least the primary estimand analysis.
Discussion
SEPRA is a pragmatic clinical trial comparing the effects of once-weekly subcutaneous semaglutide versus standard of care when used as treatment intensification, in a real-world population of adults with T2D across a variety of practice settings in the USA. The trial was self-assessed using the PRECIS-2 tool and scored 4–5 in all nine domains suggesting a highly pragmatic study. The participants recruited demonstrated high variability across specific baseline characteristics (including a wide distribution of baseline HbA1c values and BMI). Recruitment challenges were mitigated using different approaches and, encouragingly, most participants screened were enrolled, adding to the generalizability of study findings to the wider US population.
Several clinical trials described as pragmatic have been completed to date in T2D, including the EXSCEL study, which assessed the effect of exenatide once weekly versus placebo on CV outcomes in 14 752 participants34 and the Grading of Recommendations, Assessment, Development and Evaluations (GRADE) study, in which 5047 participants on metformin monotherapy were randomized to add-on glimepiride, sitagliptin, liraglutide, or insulin glargine.35 Highly pragmatic clinical trials are typically carried out in real clinical practice following usual care and build on explanatory clinical trial findings by generating real-world evidence on the comparative effectiveness of an intervention in routine clinical practice.27 In contrast, highly explanatory clinical trials are designed to produce the highest level of clinical evidence available for assessing the clinical efficacy of an intervention and typically recruit a very specific patient group.36 A continuum exists between explanatory and pragmatic clinical trials.27 To quantify the degree of pragmatism of SEPRA, the PRECIS-2 tool was used and scored 4–5 across all domains.27
A key strength of this study is the relatively high level of pragmatism, as illustrated by the retrospective assessment using the PRECIS-2 tool, which enhanced real-world representativeness. The eligibility criteria were minimally restrictive and aligned with the indication for once-weekly subcutaneous semaglutide in T2D, allowing evaluation of treatment intervention in real-life daily practice in randomized participants. However, operational challenges resulted in a slower enrollment rate than originally projected. To mitigate this, the eligibility criteria were amended to include: (i) participants with T2D receiving two or fewer oral antidiabetes medications (excluding oral semaglutide), rather than metformin alone and (ii) participants with any health plan with pharmacy benefits, instead of Anthem-affiliated plans only. The changes were judged to increase the pragmatism of the eligibility domain by broadening the study population to become more heterogenous and more representative of a real-world setting. Another key operational challenge was the emergence of the COVID-19 pandemic that led to a national lockdown in the USA and reduced the study recruitment rate. An operational decision was made to amend the exclusion criteria to allow enrollment of study sites with prior experience of semaglutide research. This increased the recruitment rate and also broadened the setting and recruitment domains within the study, making the study more explanatory (ie, less pragmatic). While the overall effect of the COVID-19 pandemic is unknown at this time, it may have impacted follow-up in some patients. While protocol amendments may thus have affected the level of pragmatism of some of the domains in potentially either direction, we believe the overall score remains the same.
There are some limitations to note due to the pragmatic design. Enrolling sites that were mainly non-research, routine-care settings maximized the pragmatism of the setting domain but required provision of training on clinical trial procedures (ie, treatment randomization, data entry, and query resolution). The open-label design could encourage participants to be more compliant with treatment; however, the minimal protocol-mandated visits and assessments may reduce adherence compared with highly explanatory clinical trials. We also note that although study visits have been kept to a minimum, treating study physicians were required to capture data that would not be captured during usual visits (eg, PROs).
The study design sought to ensure equal access to the study medication regardless of participants’ insurance status. External validity may increase if there is no reimbursement of participants’ out-of-pocket costs, while internal validity may decrease if these costs differ between arms, which could affect participants’ behavior, including adherence and persistence to medication. Thus, to balance internal and external validity, the equalized out-of-pocket cost for randomized treatment was applied. Finally, we anticipate that claims data will not be available for all participants.
In summary, SEPRA is a highly pragmatic study that has enrolled a study population with a broad range of demographic and clinical characteristics. The study is ongoing and will provide data on the effects of once-weekly subcutaneous semaglutide in a real-world population to bridge the gap between clinical trial evidence and clinical practice.
Data availability statement
Data will be shared with bona fide researchers who submit a research proposal approved by the independent review board. Individual patient data will be shared in data sets in a de-identified and anonymized format. Data will be made available after research completion and approval of the product and product use in the EU and the USA. Information about data access request proposals can be found at novonordisk-trials.com.
Ethics statements
Patient consent for publication
Ethics approval
All study activities were conducted in accordance with Good Clinical Practice Guidelines. All study activities were approved by Quorum and Advarra (institutional review board approval numbers: 33226; Pro0034694, respectively). Study personnel at physician sites were provided training on the study protocol, the Informed Consent Form, data collection, and data entry to ensure both the protection of study participants as well as the scientific integrity of the study. Site monitoring was conducted by HealthCore staff. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We gratefully acknowledge the participants, investigators, and study-site staff who were involved in the conduct of the trial. We also thank the trial managers at Novo Nordisk and HealthCore, Mardi (Margaret) Mazzeo and Susan Price. Medical writing and editorial support were provided by Beth Degg, of Axis, a division of Spirit Medical Communications Group (and were funded by Novo Nordisk), under direction of the authors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors JBB, JM, BPS, EZ, MC: design and concept, acquisition, analysis or interpretation of data, and critical revision of the manuscript for intellectual content. HNC: design and concept, acquisition, analysis or interpretation of data, drafting of the manuscript, and critical revision of the manuscript for intellectual content. BJH: acquisition, analysis or interpretation of data, critical revision of the manuscript for intellectual content, and statistical analysis. VW: design and concept, acquisition, analysis or interpretation of data, drafting of the manuscript, and critical revision of the manuscript for intellectual content. BH and VW had full access to all data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis and VW takes responsibility for the overall content as the guarantor. All authors read and approved the final manuscript.
Funding This study was funded by Novo Nordisk, Plainsboro, New Jersey, USA.
Competing interests BPS received support for the present manuscript from Novo Nordisk as a full-time employee. Holds stock or stock options for Novo Nordisk as an employee and Bristol Myers Squibb as a former employee. JBB received support for the present manuscript from Novo Nordisk, funded the study, writing support, and fees to my institution. Received grants from NIH, Dexcom, NovaTarg, Novo Nordisk, Sanofi, Tolerion, and vTv Therapeutics for clinical trials. Consulting fees (personal) from Alkahest, Altimmune, Anji, AstraZeneca, Bayer, Biomea Fusion, Boehringer-Ingelheim, CeQur, Cirius Therapeutics, Dasman Diabetes Center (Kuwait), Eli Lilly, Fortress Biotech, GentiBio, Glycadia, Glyscend, Janssen, MannKind, Mediflix, Medscape, Mellitus Health, Moderna, Pendulum Therapeutics, Praetego, ReachMD, Sanofi, Stability Health, Valo, Zealand Pharma. Consulting fees (funds to institution) from Novo Nordisk. Support for attending meetings and/or travel from AstraZeneca, Boehringer-Ingelheim, Dasman Diabetes Institute, Eli Lilly, Novo Nordisk, Zealand in the context of consulting. Participation on a data safety monitoring board or advisory board from Alkahest, Altimmune, Anji, AstraZeneca, Bayer, Biomea Fusion, Boehringer Ingelheim, CeQur, Cirius Therapeutics, Eli Lilly, GentiBio, Glycadia, Glyscend, Janssen, MannKind, Mellitus Health, Moderna, Pendulum Therapeutics, Praetego, Sanofi, Stability Health, Valo, Zealand Pharma. Leadership or fiduciary role in other board, society, committee, or advocacy group, paid or unpaid from Association for Clinical and Translational Science. Holds stock or stock options for Glyscend, Mellitus Health, Pendulum Therapeutics, PhaseBio, Praetego, and Stability Health. Is a member of the Editorial Board of BMJ Open Diabetes Research & Care. MJC received support for the present manuscript from HealthCore. Employer HealthCore received funding from Novo Nordisk to perform the study services. Holds stock or stock options for Elevance Health as a stockholder in ELV, which is the parent company of HealthCore. EWZ received medical writing support from Spirit Medical Communications. Support for attending meetings and/or travel from Novo Nordisk as an employee. Other financial or non-financial interests from Novo Nordisk as an employee. BJH received support for the present manuscript from Novo Nordisk, as sponsor of the SEPRA trial, contracted with employer, HealthCore, to serve as the data coordinating center for the trial, performing all data collection and analysis, and supporting manuscript writing activities. HNC received support for the present manuscript from Novo Nordisk as an employee. Holds stock or stock options for Novo Nordisk. VW received support for the present manuscript from HealthCore. Employer HealthCore received funding from Novo Nordisk to perform the study services. Holds stock or stock options for Elevance Health as a stockholder in ELV, which is the parent company of HealthCore.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.