Article Text

Abstract
Background Glucagon-like peptide-1 (GLP-1) receptor activation delays the progression of diabetic nephropathy (DN) in rodents. The NOD-like receptor 3 (Nlrp3) inflammasome plays an important role in DN. Dipeptidyl peptidase-4 inhibitors (DPP4I) inhibit the degradation of endogenous GLP-1 and various other active substances. We assessed whether DPP4I attenuates diabetes-induced activation of the inflammasome and progression of DN in mice with type 2 diabetes mellitus (T2DM) and type 1 diabetes mellitus (T1DM).
Methods BTBR (T2DM), Akita (T1DM) and their matched non-diabetic control (wild-type (WT)) mice received 8-week treatment with Saxagliptin (Saxa) or vehicle.
Results Kidney weight and kidney/body weight ratio increased in the BTBR and Akita mice compared to their WT mice. Saxa attenuated these changes in the BTBR, but not in the Akita mice and had no effect in the WT mice. Serum blood urea nitrogen and creatinine significantly increased in the BTBR and Akita mice. Saxa attenuated the increase in the BTBR and Akita mice. Saxa improved glycemic control in the BTBR mice, but had no effect on glucose levels in the Akita and WT mice. Serum C reactive protein, tumor necrosis factor α (TNFα), interleukin (IL)-1β, IL-6 and IL-18 were significantly higher in the BTBR and Akita mice than in the WT mice. Saxa attenuated the increase in the BTBR and Akita mice. Kidney and adipose protein levels of apoptosis-associated speck-like protein 1, NLRP3, TNFα and Caspase-1 were higher in the BTBR and Akita mice than in the WT mice. Saxa reduced the levels in both types of diabetic mice.
Conclusions Saxa attenuated diabetes-induced activation of the inflammasome and progression of DN. As Saxa did not affect glucose levels in the Akita mice, these effects are independent of glucose lowering.
- Experimental Diabetes
- Nephropathy
- Dipeptidyl Peptidase IV
- Inflammation and Complications
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
Serum blood urea nitrogen and creatinine significantly increased in the BTBR and Akita mice. Saxa attenuated the increase in the BTBR and Akita mice.
Serum C reactive protein, tumor necrosis factor α (TNFα), interleukin (IL)-1β, IL-6 and IL-18 were significantly higher in the BTBR and Akita mice than in the WT mice. Saxa attenuated the increase in the BTBR and Akita mice.
Kidney and adipose protein levels of apoptosis-associated speck-like protein 1, NLRP3, TNFα and Caspase-1 were higher in the BTBR and Akita mice than in the WT mice. Saxa reduced the levels in both types of diabetic mice.
Introduction
According to the National Diabetes Statistics Report 2014, in 2012, 29.1 million Americans (9.3% of the population) had diabetes mellitus (DM). Among Americans ≥65 years of age, 25.9% (11.8 million people) had DM. In 2013, a total of 1 469 000 new cases of diagnosed DM among adults (aged 18–79 years) were reported in the USA. DM is the leading cause of kidney failure (44% of all new cases in 2011). In 2011, a total of 228 924 American people of all ages with kidney failure secondary to DM were treated with chronic dialysis or underwent kidney transplant (http://www.cdc.gov/diabetes/data/index.html).1 It is well known that inhibition of the renin–angiotensin system by either ACE inhibitors or angiotensin type 1 receptor blockers delays, but cannot completely prevent, the progression of diabetic nephropathy (DN).2 No other established intervention has been reported to prevent and/or ameliorate DN beside the indirect effects of lowering glucose levels.3
Glucagon-like peptide-1 (GLP-1) augments glucose-dependent insulin secretion.4 GLP-1 receptor is also expressed in endothelial cells and the kidney.5–9 GLP-1 has anti-inflammatory actions. GLP-1 receptor activation inhibits the expression of vascular cell adhesion molecule-1 and tumor necrosis factor α (TNFα)10 in endothelial cells, and NFκB and intercellular adhesion molecule-1 (ICAM-1) in the kidney.11 Moreover, it was suggested that GLP-1 receptor activation increases intracellular cyclic adenosine monophosphate (cAMP) with subsequent activation of protein kinase A and downstream inactivation of phospho-c-Raf/phosphor-Erk 1/2, which are activated by angiotensin II.12 This attenuates angiotensin II-induced upregulation of plasminogen activator inhibitor-1 (PAI-1), CD68 and CXCL2 levels.12 Several studies have suggested that GLP-1 receptor activation ameliorates DN in rodents.11 ,13–17 However, Mima et al12 suggested that the protective effect of GLP-1 is attenuated by hyperglycemia and free fatty acids, as hyperglycemia and high levels of free fatty acids upregulate protein kinase C β that decreases GLP-1 receptor expression and activates the proinflammatory phosphor-c-Raf.
Dipeptidyl peptidase-4 inhibitors (DPP4I) inhibit the degradation of endogenous GLP-1 and, therefore, augment its circulation levels.18 However, DPP4Is also prevent the degradation of other non-GLP-1 substrates such as glucose-dependent insulinotropic peptide (GIP), B type natriuretic peptide, substance P, neuropeptide Y, peptide YY, bradykinin and stromal-derived factor 1α (SDF-1α) and, thus, could have effects that differ from that of pure GLP-1 receptor activation.18 The effects of DPP4I on the progression of DN have been less described. It was reported that DPP4I-induced natriuresis in non-diabetic mice was independent of the GLP-1 receptors. Moreover, in mice with type 2 DM (T2DM, db/db), DPP4I (alogliptin), in contrast to the GLP-1 receptor agonist, Exendin-4, failed to inhibit renal fluid and sodium reabsorption.19 On the other hand, Kodera et al20 reported that DPP4I with PKF275-055 attenuated inflammation, NFκB activation and delayed the progression of DN in rats with type 1 DM (T1DM, streptozotocin-induced).
The NOD-like receptor 3 (Nlrp3) inflammasome is an interleukin (IL)-1β family cytokine-activating protein complex. The inflammasome plays an important role in the inflammation associated with T2DM21–24 and various forms of renal injury.25–27 Nlrp3 interaction with apoptosis-associated speck-like protein (ASC) leads to activation of caspase-1, with subsequent increase in the production of proinflammatory cytokines.23 ,25 ,26 It is reported that glyburide (sulfonylurea) prevents activation of inflammasome and delays lipopolysaccharide-induced lethality in mice.28 The Nlrp3 inflammasome is involved in DN in the rat and could be inhibited with allopurinol.29 ,30 An in vitro study suggested that DPP4I represses the Nlrp3 inflammasome and IL-1β expression in macrophages.31 However, the effects of GLP-1 and or DPP4I treatment on the activation of the Nlrp3 inflammasome in the diabetic kidney has not been reported. We investigated the effects of Saxagliptin (Saxa) on the development of DN and Nlrp3 inflammasome activation using a mouse model of T1DM and T2DM.
Methods
Treatment
Male BTBR ob/ob mice {BTBR.V(B6)-Lepob/WiscJ}, male C57BL/6 mice (wild-type (WT) control for the BTBR), male AKITA mice {C57BL/6NJcl-Ins2Akita} and male C57BL/6NJcl mice (WT control for the Akita) were purchased from Jackson Laboratory.32 ,33 At an age of 8 weeks, mice received (1) C57BL/6+vehicle; (2) BTBR+vehicle; (3) BTBR+Saxa; (4) C57BL/6NJcl+vehicle; (5) C57BL/6NJcl+Saxa; (6) AKITA+vehicle and (7) AKITA+Saxa. As the genetic background of the C57BL/6 and C57BL/6NJcl is considered close enough, we omitted the group of C57BL/6+Saxa to look for the effects of Saxa in non-diabetic animals. Saxa (10 mg/kg/day) was mixed with the normal chow (276.7 mg/kg of chow) and was administered for 8 weeks. After completion of treatment, body weight was measured and fasting glucose was determined using a glucometer (One Touch, Wako Diagnostics, Richmond, Virginia, USA).
All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication number 85-23, Revised 1996). The protocol was approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch.
Glucose tolerance test
Intraperitoneal glucose tolerance test (IGTT) was carried out after 8-week treatment. Mice had fasted for 4 h prior to IGTT. Glucose (1 g/kg of body weight) was injected intraperitoneally. Blood samples were collected from the tail vein before (0 min) and 30, 60, 90 and 120 min after the glucose administration. Blood glucose levels were determined using glucometer.
Enzyme-linked immunosorbent assays
After anesthetizing the animals with ketamine (20 mg/kg) and xylazine (4 mg/kg), blood was collected from the left ventricle. Plasma was collected using EDTA as an anticoagulant. Samples were centrifuged for 15 min at 1000×g at 2–8°C within 30 min of collection. C reactive protein (CRP), IL-1β, IL-18, IL-6 and TNFα levels in serum were determined by ELISA (R&D Systems, Minneapolis, Minnesota, USA).
Kidney function
The weights of kidneys (KW) and the ratio KW/body weight were determined. Blood urea nitrogen (BUN) and creatinine were estimated as markers of renal dysfunction.
Renal morphology assessment
Samples were fixed in 4% paraformaldehyde and embedded in paraffin wax. Five-micrometer thick sections were stained for routine histopathological diagnosis with H&E. Periodic acid of Schiff (PAS) staining was performed to evaluate the interstitium of the kidney. High-quality digital images suitable for quantitative analysis were captured from PAS-stained section using light microscope (Olympus DP71) at a magnification of ×400. The glomerular tuft area was measured (at least 50 samples) using Image J software. The mean glomerular area was obtained. Then PAS-positive material in each glomerulus was quantified and expressed as a percentage of the glomerular tuft area (fractional mesangial area).34 ,35
Western blot analysis
Renal cortex and adipocyte levels of TNFα, NLRP3, ASC1 and Caspase-1 were evaluated by western blot. Samples were homogenized in lysis buffer (in mM): 25 Tris–HCl (pH 7.4), 0.5 EDTA, 0.5 EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), 1 phenylmethylsulfonyl fluoride, 1 dithiothreitol, 25 NaF, 1 Na3VO4, 1% Triton X-100, 2% SDS and 1% protease inhibitor cocktail. The lysate was centrifuged at 10 000×g for 15 min at 4°C and supernatants were collected. Protein concentration was determined by the Bradford method. Protein (50 µg) was fractionated by SDS-PAGE (4–20% polyacrylamide gels) and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, Massachusetts, USA).
After blocking, membranes were incubated with primary antibodies anti-TNFα, anti-ASC1, anti-caspase-1 (Santa Cruz Biotechnology, USA), anti-NLRP3 (R&D System, USA) or anti-β-actin (Sigma-Aldrich, USA) overnight at 4°C. The membranes were then washed and incubated with secondary membrane horseradish peroxidase (HRP)-conjugated antibodies for 1 h at room temperature. Bound antibodies were detected using the chemiluminescent substrate (NEN Life Science Products, Boston, Massachusetts, USA). The protein signals were quantified with an image-scanning densitometer, and the strength of each protein signal was normalized to the corresponding β-actin signal. Data are expressed as per cent relative the expression in the control group (WT+vehicle).
Reverse transcription polymerase chain reaction
Total RNA from isolated mouse renal and adipose tissue was extracted using TRIzol reagent (Invitrogen, Carlsbad, California, USA) according to the protocol per manufacturer's instruction. Two micrograms of total RNA from each sample was reverse transcribed into cDNA and equal amounts of the reverse transcriptional products were subjected to PCR amplification. The evaluation of mRNA expression of TNFα, NLRP3, ASC, Caspase-1, IL1β, IL6 and IL18 was performed by using the primers reported in table 1. The Ct (threshold cycle) is defined as the number of cycles required for the fluorescence signal to exceed the detection threshold. Expression of the gene relative to the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was calculated as the difference between the threshold values of these two genes (2−ΔCt). Melting curve analysis was performed during real-time PCR to analyze and verify the specificity of the reaction. The values are given as the means±SE of four independent experiments. Each sample was analyzed in triplicate and normalized by GAPDH.
Primer sequences for real-time RT-PCR
Results
Body weight (figure 1A) of the BTBR mice was significantly higher than the WT mice (p<0.001). Body weight in the BTBR+Saxa group was significantly higher than in the WT group, but lower than in the BTBR group (p<0.001). Body weight was comparable between the WT and Akita mice. Saxa had no effect on body weight in the WT and Akita mice.

(A) Body weight (g). T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=5 per group. (B) Kidney weight (g). T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=5 per group. *p<0.001 vs WT. (C) Kidney/body weight ratio (×1000). T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=5 per group. *p<0.001 vs WT. (D) Serum BUN. T2DM: n=5 per group. *p<0.009 vs WT; †p<0.001 vs BTBR. T1DM: n=5 per group. *p<0.031 vs WT; †p<0.001 vs Akita. (E) Serum creatinine. T2DM: n=5 per group. *p<0.003 vs WT; †p<0.001 vs BTBR. T1DM: n=5 per group. *p<0.035 vs WT; †p<0.001 vs Akita. T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; WT, wild-type.
Kidney weight (figure 1B) and the kidney weight/body weight ratio (figure 1C) were significantly higher in the BTBR than in the WT group (p<0.001 for both). Kidney weight and the kidney/body weight ratio were significantly lower in the BTBR+Saxa group compared to the BTBR group (p<0.001 for both). Kidney weight and the kidney/body weight ratio were significantly higher in the Akita than in the WT mice (p<0.001 for both). Saxa had no effect on these parameters in the WT and the Akita mice.
Serum BUN (figure 1D) and creatinine (figure 1E) were significantly higher in the BTBR mice than in the WT mice (p<0.001). Saxa attenuated the increase in the BTBR mice (p<0.001 vs BTBR; p<0.009 vs WT). Serum BUN and creatinine were significantly higher in the Akita mice than in the WT mice (p<0.001 for both). Saxa had no effect on BUN and creatinine in the WT mice; however, it attenuated the increase in BUN and creatinine in the Akita mice (p<0.001 for both vs Akita).
Fasting serum glucose was significantly higher in the BTBR (339.0±10.0 mg/dL) than in the WT mice (107.7±2.9 mg/dL; p<0.001). Saxa significantly reduced glucose levels in the BTBR mice (163.8±5.9 mg/dL; p<0.001 vs BTBR and WT) (n=6 per group). Serum fasting glucose was significantly higher in the Akita mice (404.0±10.0 mg/dL) than the WT mice (122.4±3.8 mg/dL; p<0.001). In contrast to the effect in the BTBR mice, Saxa had no significant effect on fasting serum glucose in the WT (118.0±3.9 mg/dL; p=0.999) or the Akita (405.0±11.8 mg/dL; p=1.0) mice (n=5 per group).
Glucose levels during all time points after glucose loading were significantly higher in the BTBR mice than in the WT mice (figure 2A). Saxa significantly reduced glucose levels in the BTBR mice.

(A) T2DM: serum glucose levels during glucose tolerance test (n=6 per group). Two-way repeat measures ANOVA shows a significant group effect (p<0.001) and time effect (p<0.001) (p<0.051 for the group×time interaction). (B) T1DM: serum glucose levels during glucose tolerance test (n=x per group). Two-way repeat measures ANOVA shows a significant group effect (p<0.001) and time effect (p<0.001) (p<0.001 for the group×time interaction). ANOVA, analysis of variance; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; WT, wild-type.
Glucose levels during all time points after glucose loading were significantly higher in the Akita mice compared to the WT mice (p<0.001) (figure 2B). Saxa had no effect on glucose levels in the WT (p=0.958) and the Akita mice (p=0.975).
Serum CRP levels significantly increased in the BTBR and Akita mice compared to their matched WT mice (figure 3A). Saxa attenuated the increase in the BTBR mice and completely blocked it in the Akita mice. Saxa had no effect on CRP levels in the WT mice. Serum TNFα (figure 3B), IL-1β (figure 3C), IL-6 (figure 3D) and IL-18 (figure 3E) were significantly higher in the BTBR and Akita mice compared to the WT non-diabetic mice. Saxa significantly attenuated the levels in the BTBR mice and the Akita mice, whereas Saxa had no effect in the WT mice.

(A) Serum CRP levels. T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=5 per group. *p<0.001 vs WT; †p<0.001 vs Akita. (B) Serum TNFα. T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs Akita. (C) Serum IL-1β T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs Akita. (D) Serum IL-6. T2DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs BTBR. T1DM: n=6 per group. *p<0.001 vs WT; †p<0.001 vs Akita. (E) Serum IL-18. T2DM: n=6 per group. *p<0.03 vs WT; †p<0.001 vs BTBR. T1DM: n=6 per group. *p<0.003 vs WT; †p<0.001 vs Akita. CRP, C reactive protein; IL, interleukin; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TNFα, tumor necrosis factor α; WT, wild-type.
In the kidney, levels of ASC1, NLRP3, TNFα and Caspase-1 were significantly higher in the diabetic mice than in the WT mice (figure 4). Saxa had no effect on the expression of these proteins in the WT mice. In contrast, it attenuated the expression of ASC1, NLRP3 and TNFα in the BTBR and Akita mice. In addition, Saxa attenuated the increase in Caspase-1 expression in the BTBR mice and completely blocked the increase in the Akita mice.

Immunoblotting samples (A and F) and densitometric analysis of ASC1 (B and G), NLRP3 (C and H), TNFα (D and I) and Caspase-1 (E and J) expression in the kidney of the WT, BTBR and Akita mice without and with Saxa treatment. N=4 per group. *p<0.02 vs WT; †p<0.03 vs BTBR or Akita mice. ASC1, apoptosis-associated speck-like protein 1; TNFα, tumor necrosis factor α; WT, wild-type.
Similar results were obtained in the adipose tissue (figure 5). T1DM and T2DM increased ASC1, NLRP3, TNFα and Caspase-1 levels. Saxa attenuated the increase in ASC1, NLRP3 and TNFα levels. Saxa attenuated the increase in Caspase-1 in BTBR mice and completely blocked the increase in the Akita mice.

Immunoblotting samples (A and F) and densitometric analysis of ASC1 (B and G), NLRP3 (C and H), TNFα (D and I) and Caspase-1 (E and J) expression in the adipose tissue of the WT, BTBR and Akita mice without and with Saxa treatment. N=4 per group. *p<0.002 vs WT; †p<0.025 vs BTBR or Akita mice, respectively. ASC1, apoptosis-associated speck-like protein 1; TNFα, tumor necrosis factor α; WT, wild-type.
RT-PCR confirmed the results of the immunoblotting and ELISA. T1DM and T2DM increased mRNA levels of ASC, NALP3, TNFα, IL-1β, IL-6 and IL-18 in the kidney (figure 6). Saxa decreased the levels in the BTBR and Akita mice.

mRNA levels of ASC, NALP3, TNFα, IL-1β, IL-6 and IL-18 in the kidney of WT (control) versus BTBR mice (left column) or WT (control) versus Akita mice (right column). N=4 per group. BTBR: *p<0.025 vs control; †p<0.045 vs BTBR. Akita: *p<0.009 vs control; †p<0.005 vs Akita. ASC, apoptosis-associated speck-like protein; IL, interleukin; TNFα, tumor necrosis factor α; WT, wild-type.
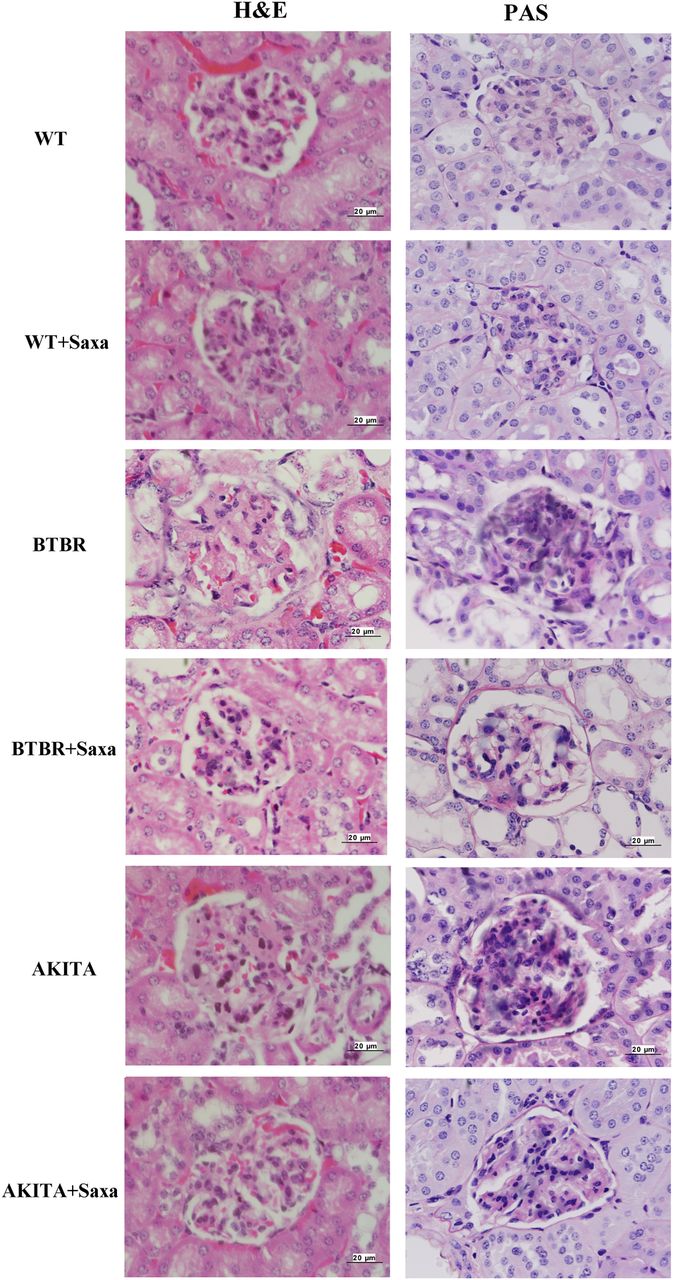
H&E and PAS reagent staining of the kidneys shows glomerular hypertrophy and mesangial expansion in the BTBR and the Akita mice (figure 7). Glomerular area was 3679±354 µm2 in the WT mice. Glomerular area increased to 9178±695 µm2 in the BTBR mice (p<0.001 vs WT). Saxa attenuated the increase in glomerular area in the BTBR mice (5369±339 µm2; p<0.001 vs BTBR; p=0.067 vs WT). Likewise, glomerular area increased in the Akita mice (9823±744 µm2; p<0.001 vs WT). Saxa attenuated the increase in glomerular area in the Akita mice (5095±321 µm2; p<0.001 vs Akita; p=0.216 vs WT). Saxa had no effect on glomerular area in the WT mice (3262±304 µm2; p=0.990 vs WT). The fractional mesangial area was 5.8±0.4% in the WT mice. It increased to 22.5±1.0% in the BTBR mice (p<0.001 vs WT). Saxa attenuated the increase in the BTBR mice (11.1±0.6%; p<0.001 vs BTBR; p<0.001 vs WT). The fractional mesangial area also increased to 27.3±1.7% in the Akita mice (p<0.001 vs WT). Saxa attenuated the increase in the Akita mice (12.9±1.0%; p<0.001 vs Akita; p=0.001 vs WT), whereas it had no effect in the WT mice (5.1±0.4%; p=0.998 vs WT). Thus, Saxa attenuated the increase in glomerular size and mesangial expansion in the BTBR and Akita mice, without a notable effect in the WT mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Representative pictures of H&E and PAS reagent staining of the kidney (original magnification ×400). PAS, periodic acid of Schiff; WT, wild-type.
Discussion
In the current study, we found that 8-week Saxa treatment significantly attenuated DM-induced activation of the NLRP3 inflammasome (figures 4 and 5), and significantly reduced the serum levels of CRP, TNFα, IL-1β, IL-18 and IL-6 (figure 3). Furthermore, Saxa delayed the progression of DN in mice with T2DM, as well as T1DM. Saxa attenuated DM-induced increase in absolute kidney weight and kidney/body weight ratio in the BTBR mice (figure 1), the increase in serum BUN and creatinine in the BTBR and Akita mice (figure 2) and the morphologic changes associated with DN (glomerular hypertrophy and mesangial expansion) in the BTBR and the Akita mice (figure 7).
As expected, Saxa decreased fasting serum glucose levels and improved glucose tolerance after glucose loading in the BTBR mice (a T2DM model), but not in the Akita mice (T1DM model) (figure 2). Thus, the beneficial effects of Saxa on the inflammasome and DN were (at least partial) independent of improvements in glucose tolerance given that Saxa had similar beneficial effects in BTBR and Akita mice, except on the kidney weight and the kidney/body weight ratio.
Several previous studies have suggested that GLP-1 receptor agonists ameliorate DN in rodents with T2DM13 as well as with T1DM.11 ,14 ,17 Park et al13 reported that 8-week treatment with exendin-4, a GLP-1 receptor agonist, delayed the progression of DN in db/db mice. They suggested that the beneficial effects are related to improvement in the metabolic anomalies due to better glycemic control and due to increased GLP-1 receptor expression in the kidney.13 On the other hand, Hendarto et al14 reported that in streptozotocin-induced T1DM rats, liraglutide (a GLP-1 receptor agonist) reduced oxidative stress via a cAMP/protein kinase A-mediated inhibition of renal NAD(P)H oxidases, transforming growth factor-β (TGF-β) and fibronectin. The protective effects were blocked by H89, a protein kinase A inhibitor, and SQ22536, an adenylate cyclase inhibitor. Kodera et al11 reported that 8-week exendin-4 reduced macrophage infiltration and decreased ICAM-1, TGF-β and type IV collagen levels in the kidney of rats with streptozotocin-induced T1DM, without affecting glycemic control. On the other hand, Ojima et al found that 2-week treatment with exendin-1 suppressed protein arginine methyltransfetase-1 (PRMT-1, an enzyme that mainly generates asymmetric dimethylarginine) levels in the kidney of streptozotocin-induced T1DM rats. In addition, exendin-1 decreased the production of advanced glycation end products (AGEs) and their receptor (RAGE), decreased ICAM-1 and monocyte chemoattractant protein 1 (MCP-1) levels and attenuated reactive oxygen species generation without affecting glycemic control.17 An in vitro study confirmed that GLP-1 inhibits RAGE gene expression and reactive oxygen species generation and MCP-1 levels in human cultured renal mesangial cells.16 Moreover, using a similar in vitro model, it was shown that GLP-1 receptor activation blocked the angiotensin-II-induced mesangial cell injury. The effect was dependent on protein kinase A activation with downstream inhibition of reactive oxygen species generation, NFκB, ICAM-1 and PAI-1 upregulation.15 Thus, collectively, the previous studies have suggested a cAMP/protein kinase A-mediated reduction in reactive oxygen species generation and inflammation. It was reported that glyburide prevents Nlrp3 inflammasome activation in vitro;28 however, the effects of GLP-1 receptor activation on the Nlrp3 inflammasome has not been reported before. The inflammasome plays an important role in the inflammation associated with T2DM21–24 and various forms of renal injury.25–27
There are scant data on the effects of DPP4Is on the progression of DN in T2DM and no data on the activation of the Nlrp3 inflammasome. In addition to preventing the degradation of endogenous GLP-1, DPP4Is prevent the degradation of numerous substrates such as GIP, B type natriuretic peptide, substance P, neuropeptide Y, peptide YY, bradykinin and SDF-1α and, thus, could have effects that differ from that of pure GLP-1 receptor activation.18 Kanasaki et al found that linagliptin ameliorated kidney fibrosis in streptozotocin-induced diabetic mice. They observed that levels of microRNA 29s were reduced in the kidneys of the diabetic mice and that linagliptin restored their levels; however, the exact signaling pathway for restoring microRNA 29 levels was not specified.36 Kodera et al20 reported that 8-week DPP4I with PKF275-055 attenuated inflammation, NFκB activation and delayed the progression of DN in rats with streptozotocin-induced T1DM. Nakashima et al37 found that linagliptin ameliorated renal damage in rats with streptozotocin-induced T1DM. Linagliptin did not affect glycemic control, but significantly reduced AGEs RAGE.37 Matsui et al38 reported that the development of DN was delayed in DPP-4-deficient rats than in WT rats with streptozotocin-induced T1DM. As there were no differences in glucose levels and lipid parameters between the WT and the DPP4-deficient mice, they concluded that the protective effect was independent of glycemic control. They suggested that the protective effect was due to blocking of AGEs and RAGE.38 In contrast, Rieg et al19 found that in db/db mice with T2DM, alogliptin (a DPP4I), in contrast to Exendin-4, failed to inhibit renal fluid and sodium reabsorption. Although an in vitro study suggested that DPP4I represses the Nlrp3 inflammasome and IL-1β expression in macrophages,31 our study is the first to show that the DPP4I Saxa attenuated Nlrp3 inflammasome activation in vivo in mice with T2DM, as well as T1DM, suggesting that at least part of the beneficial effect is independent of improvements in glucose tolerance.
The Nlrp3 inflammasome complex activates caspase-1 that regulates the activation and secretion of IL-1β and IL-18 from cells.21 ,23 ,25 ,26 The inflammatory cytokines TNFα, IL-1β, IL-6 and IL-18 have crucial roles in mediating the glomerular and tubulointerstitial injury induced by diabetes.39 ,40 Here we are showing that in addition to reducing the levels of the components of the Nlrp3 inflammasome, Saxa attenuated the increase in all these four cytokines in T2DM and T1DM models.
The exact mechanism(s) of attenuating the DM-induced activation of the Nlrp3 inflammasome by Saxa have not been elucidated. Previous studies suggested that the anti-inflammatory effects of GLP-1 receptor activation are dependent on cAMP-induced protein kinase A activation.14 ,15 Our group has previously shown that GLP-1 receptor agonists and DPP4Is increased the production of 15-epi-lipoxin A4, an arachidonic acid mediator with potent anti-inflammatory and inflammation resolution properties via activation of protein kinase A and 5-lipoxygenase.41 ,42 15-Epi-lipoxin A4 suppressed activation of the inflammasome.43 ,44 Thus, inhibition of the inflammasome in our models might be explained by 15-epi-lipoxin A4. However, protein kinase A has numerous other targets. Further studies are needed to establish the role of 15-epi-lipoxin A4 in mediating the protective effect of DPP4Is. It has been suggested that protein kinase A inactivates phospho-c-Raf/phosphor-Erk 1/2, which are activated by angiotensin II.12 This attenuates angiotensin II-induced upregulation of PAI-1, CD68 and CXCL2 levels.12
A recent study suggested that in streptozotocin-induced type 1 diabetic mice, the levels of endothelial DPP4, integrin β1 and phosphor-integrin β1 were all higher than in non-diabetic mice.45 The DPP4I linagliptin suppressed endothelial levels of DPP4, integrin β1 and phospho-integrin β1, reduced plasma cystatin C levels and reduced kidney fibrosis. Suppression of DPP4 by siRNA was associated with suppression of integrin β1 and vice versa. Knockdown of either integrin β1 or DPP4 resulted in the silencing of TGF-β2-induced TGF-β receptor heterodimer formation, smad3 phosphorylation and endothelial-to-mesenchymal transition.45 The authors suggested that inhibiting DPP4 may be a therapeutic target for treating kidney fibrosis in diabetes.45 It is yet unclear whether such an interaction between integrin β1 and DPP4 affects inflammasome activation in mediating fibrosis.45
Limitations
Although it is well established that DPP4Is increase the levels of GLP-1, in the current study, we did not measure tissue and serum levels of GLP-1. We have not collected urine sample to study the effect of treatment on microalbuminuria.
In conclusion, we found that Saxa attenuated the diabetes-induced activation of the Nlrp3 inflammasome and the upregulation of the proinflammatory cytokines TNFα, IL-1β, IL-6 and IL-18 in mice with T2DM and T1DM. Saxa significantly delayed the progression of DN in the two models. The fact that effects of Saxa were comparable in the Akita and BTBR mice, despite the fact that Saxa had no effects on glycemic control in the T1DM model, suggests a signaling pathway that is (at least partially) independent of glucose-lowering effect of SAXA. Our important findings in rodent models should be confirmed in patients with T1DM and T2DM, as inhibition of the Nlrp3 inflammasome and reducing TNFα, IL-1β, IL-6 and IL-18 levels might have significant implications in the clinical setting for preventing DN.
References
Footnotes
YB and MB contributed equally.
Contributors YB contributed to study design, data collection and analysis, statistical analysis and writing the manuscript. MB involved in data analysis, statistical analysis and cowriting the manuscript. JQ and MB involved in data analysis, searching literature and editing the manuscript. YY contributed to study design, submitting the grant, conduction of the experiments, data collection and analysis, literature search and editing the manuscript.
Funding This work was supported by a grant from AstraZeneca to YB and YY by a research grant from the American Diabetes Association to MB (07-13-TS-04).
Competing interests YB has received research grants from AstraZeneca and Boehringer Ingelheim. He has received lecture fees from Daiichi Sankyo and AstraZeneca. MB has received research grants from AstraZeneca, Boehringer Ingelheim, Eli-Lilly and Novo Nordisk. He has received lecture fees from Takeda Pharmaceuticals and Sanofi Aventis and is a consultant to Merck and Genentech. YY has received research grants from AstraZeneca and Boehringer Ingelheim.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.