Article Text

Abstract
Objective To evaluate the risk of serious adverse events among patients with type 2 diabetes mellitus initiating saxagliptin compared with oral antidiabetic drugs (OADs) in classes other than dipeptidyl peptidase-4 (DPP-4) inhibitors.
Research design and methods Cohort studies using 2009–2014 data from two UK medical record data sources (Clinical Practice Research Datalink, The Health Improvement Network) and two USA claims-based data sources (HealthCore Integrated Research Database, Medicare). All eligible adult patients newly prescribed saxagliptin (n=110 740) and random samples of up to 10 matched initiators of non-DPP-4 inhibitor OADs within each data source were selected (n=913 384). Outcomes were hospitalized major adverse cardiovascular events (MACE), acute kidney injury (AKI), acute liver failure (ALF), infections, and severe hypersensitivity events, evaluated using diagnostic coding algorithms and medical records. Cox regression was used to determine HRs with 95% CIs for each outcome. Meta-analyses across data sources were performed for each outcome as feasible.
Results There were no increased incidence rates or risk of MACE, AKI, ALF, infection, or severe hypersensitivity reactions among saxagliptin initiators compared with other OAD initiators within any data source. Meta-analyses demonstrated a reduced risk of hospitalization/death from MACE (HR 0.91, 95% CI 0.85 to 0.97) and no increased risk of hospitalization for infection (HR 0.97, 95% CI 0.93 to 1.02) or AKI (HR 0.99, 95% CI 0.88 to 1.11) associated with saxagliptin initiation. ALF and hypersensitivity events were too rare to permit meta-analysis.
Conclusions Saxagliptin initiation was not associated with increased risk of MACE, infection, AKI, ALF, or severe hypersensitivity reactions in clinical practice settings.
Trial registration number NCT01086280, NCT01086293, NCT01086319, NCT01086306, and NCT01377935; Results.
- saxagliptin
- post-authorization safety study
- type 2 diabetes mellitus
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Saxagliptin, a dipeptidyl peptidase-4 inhibitor, is an oral antidiabetic drug used in combination with diet and exercise to control hyperglycemia in adults with type 2 diabetes mellitus.
Despite its widespread use in oral hypoglycemic therapy, few studies have evaluated the safety of saxagliptin in real-world settings.
What are the new findings?
Saxagliptin initiation was not associated with significantly increased incidence rates or risk of major adverse cardiovascular events, infection, acute kidney injury, acute liver failure, or severe hypersensitivity reactions.
How might these results change the focus of research or clinical practice?
These data provide no evidence for concern about an increase in risk of these outcomes from saxagliptin in real-world settings.
Introduction
Saxagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor, is an oral antidiabetic drug (OAD) used in combination with diet and exercise to control hyperglycemia in adults with type 2 diabetes mellitus. This medication is approved for use as both monotherapy and combination therapy in the USA (approved July 2009) and as combination therapy in the European Union (approved October 2009).1 2 Despite its widespread use in oral hypoglycemic therapy, few studies have evaluated the safety of saxagliptin in real-world settings. Since clinical trials are typically underpowered to detect uncommon, but potentially life-threatening, adverse reactions, postmarketing assessments are important to identify important medication-related toxicities in routine clinical practice.3
Prior to saxagliptin’s approval in the USA and European Union, we designed an observational study within USA and UK practice settings to evaluate associations between saxagliptin and five outcomes of importance to patients with type 2 diabetes mellitus receiving OAD therapy, including major adverse cardiovascular events (MACE), acute liver failure (ALF), acute kidney injury (AKI), infections, and severe hypersensitivity reactions. After saxagliptin’s approval, we prospectively collected data from 2009 to 2014 and compared the incidence rates and risk of each outcome between patients with type 2 diabetes mellitus who were new initiators of saxagliptin and those who were new initiators of OADs in classes other than DPP-4 inhibitors.
Research design and methods
Data sources
We conducted cohort studies within two UK medical record data sources (Clinical Practice Research Datalink (CPRD), The Health Improvement Network (THIN)) and two USA claims-based data sources (HealthCore Integrated Research Database (HIRD), Medicare).4 The study protocol and variables evaluated within each data source were previously described.5
Within the UK, CPRD contains electronic healthcare records for >15 million patients across 684 practices6 and THIN contains medical records for >11 million patients across 550 practices.7 8 This study analyzed the first 52 months of saxagliptin availability within the UK (2009–2014), using the March 2014 version of CPRD and the 1401 version of THIN. CPRD and THIN collect demographics, medical diagnoses and surgical procedures (recorded using Read codes), outpatient laboratory results, general practitioner-issued prescriptions, hospital admission and discharge dates, and dates and causes of death from Office for National Statistics’ death certificate data.9 10 Since some practices contribute to both CPRD and THIN, we identified and excluded patients in THIN who were also present within CPRD to avoid double-counting these individuals within analyses.11
Within the USA, HIRD is a commercial health insurance database serving 23.2 million members.12–14 We analyzed HIRD data over the first 53 months of saxagliptin availability in the USA (2009–2013). Medicare is a federal health insurance program available to Americans aged ≥65 years and those under 65 years with certain disabilities or chronic health conditions.15 We analyzed Medicare data over the first 41 months of saxagliptin availability in the USA (2009–2012) only, due to the 18-month time-lag in Medicare data availability for research. Both HIRD and Medicare contain demographic information, inpatient and outpatient medical diagnoses (recorded using International Classification of Diseases, Ninth Revision codes), surgical procedures (recorded with Current Procedural Terminology codes), and dispensed medications (recorded by National Drug Codes). The National Death Index was used to determine date and cause of death in both USA databases. To prevent double-counting patients concurrently enrolled in both USA data sources, HIRD data were only included for patients 18–64 years of age and enrollees were censored at age 65 years.
This study was approved by the UK Independent Scientific Advisory Committees for CPRD (Protocol 10_149RMn) and THIN (Protocol 11–039V), Quorum Review Institutional Review Board for HIRD, and Institutional Review Boards of the University of Pennsylvania and Rutgers University. A data use agreement was obtained from the Centers for Medicare and Medicaid Services.
Study patients
Patients were eligible if they were (1) ≥18 years, (2) newly initiated saxagliptin or an OAD in a class other than DPP-4 inhibitors (with or without additional OADs), and (3) enrolled in their data source for ≥180 days prior to initiation of saxagliptin or comparator OAD. The exposed cohort consisted of initiators of saxagliptin, prescribed as a single agent or a fixed-dose combination with metformin. The unexposed cohort consisted of initiators of OADs in classes other than DPP-4 inhibitors. Our rationale for not including initiators of other DPP-4 inhibitors in the comparator group was to ensure that we did not miss potentially important associations related to the DPP-4 class. Patients were excluded from primary analyses (though included in a sensitivity analysis) if they received insulin or glucagon-like peptide-1 (GLP-1) receptor agonists, since at the time of protocol design thorough investigations regarding the use of these therapies with saxagliptin had not been performed.
Within each data source, we selected all eligible saxagliptin initiators and a random sample of up to 10 eligible initiators of non-DPP-4 inhibitor OADs matched on age (within 5-year age groups), sex, and geographic region (ie, census region for USA data sources; country for UK data sources) to each saxagliptin initiator, to ensure sufficient sample sizes for subanalyses.
The index date was the date of first prescription/claim for saxagliptin or comparator OAD. The 180 days prior to this date represented the baseline period. Follow-up continued until study outcome, drug discontinuation (ie, no further drug claim or prescription within 30 days after the last days’ supply), non-saxagliptin DPP-4 inhibitor initiation, or end of study, whichever occurred first.
Main study outcomes
The primary outcomes were (1) hospitalization with and/or death due to MACE, (2) hospitalization with ALF, (3) hospitalization for AKI, (4) hospitalization for infection, and (5) hospitalization for severe hypersensitivity reaction. We evaluated hospitalizations for AKI, infections, and hypersensitivity reactions because these conditions are frequently hospital-acquired. Evaluating hospitalizations with these conditions would leave open opportunities for diagnostic suspicion bias and include events that were not the main reason for hospitalization.5
End points were ascertained by diagnostic coding algorithms (see online supplementary tables S1-S11). MACE was defined by a hospital diagnosis of acute myocardial infarction (AMI), acute stroke, and/or death from cardiovascular causes (ie, AMI, acute stroke, congestive heart failure, dysrhythmia, sudden death, or coronary revascularization). Hospitalization with ALF was determined by inpatient diagnosis. Hospitalization for AKI was determined by inpatient AKI diagnosis plus at least one of the following within 7 days prior to admission: (1) emergency department AKI diagnosis, (2) outpatient AKI diagnosis, or (3) available serum creatinine result (within UK data) or claim for serum creatinine or serum chemistry panel including creatinine (within USA data). Hospitalization for infection was identified by inpatient infection diagnosis plus at least one of the following within 7 days prior to admission: (1) outpatient antimicrobial prescription/claim, (2) emergency department infection diagnosis, or (3) outpatient infection diagnosis. Hospitalization for severe hypersensitivity reaction was defined by (1) inpatient diagnosis for angioedema and/or generalized urticaria plus an emergency department or outpatient diagnosis of angioedema, urticaria, or rash within 7 days prior to admission; or (2) inpatient diagnosis for anaphylaxis, Stevens-Johnson syndrome, toxic epidermal necrolysis, or other severe skin reaction.
Within each data source, we sampled patients who met each diagnostic coding algorithm, requested records from these patients from general practitioners (in the UK) and hospitals (in the USA) to enable confirmation of endpoints, had clinicians with expertise in each outcome review these records to adjudicate events using criteria we previously published,5 and calculated the positive predictive value (PPV) of the algorithms for confirmed events (see online supplementary table S12). We sought algorithms with >80% PPV to provide confidence that identified outcomes were true events. For any algorithm with <80% PPV within a data source (AKI within CPRD and THIN; infection within CPRD; ALF and severe hypersensitivity reactions within all data sources), we classified patients as having an event if (1) the outcome was confirmed by adjudication or (2) the patient met the algorithm but had no records available to confirm the event.
Data collection
Baseline data included demographic information, medical diagnoses, surgical procedures, and medications commonly prescribed in type 2 diabetes (table 1). We ascertained prior OAD use within the 180 days preceding the index date. Patients were considered to have ‘switched to’ the index drug if they were prescribed/dispensed an OAD within the 90 days prior to their index date but this drug was not prescribed/dispensed in the 90 days after that date. Patients were considered to have ‘added on’ their index drug to their OAD therapy if they continued to receive the same OADs within the 90 days prior to and 90 days after their index date.
Demographic characteristics of patients with type 2 diabetes mellitus within the UK in the Clinical Practice Research Datalink (2009–2014) and The Health Improvement Network (2009–2014)
Within the UK data sources, we collected the most recent hemoglobin A1c result prior to the index date, smoking status, and obesity (body mass index >30 kg/m2). Within the USA data sources, we collected the number of claims for hemoglobin A1c tests recorded in the baseline period since laboratory results were not available for all enrollees within these data.
Statistical analysis
Within each data source, we compared characteristics between saxagliptin and comparator OAD initiators. Patients who had a study end point during the baseline period were excluded from analyses of that outcome. Unadjusted incidence rates of outcomes were calculated by cohort in each data source.
Because of the many potential confounders relative to the number of outcomes, we used propensity scores to control for confounding.16 Propensity scores were developed within each data source using logistic regression, incorporating measured potential predictors of saxagliptin as independent variables and saxagliptin exposure as the dependent variable. We excluded patients from the saxagliptin cohort whose propensity score exceeded the maximum or minimum values in the comparator OAD cohort (trimmed the tails). All variables in tables 1 and 2 were included in propensity score models.
Demographic characteristics of patients with type 2 diabetes mellitus within the USA in Medicare (2009–2012) and the HealthCore Integrated Research Database (2009–2013)
Cox regression was used to determine HRs with 95% CIs of outcomes in saxagliptin versus other OAD initiators, adjusting for propensity score, prior OAD therapy, quarter of observation, and geographic region. We adjusted for, rather than stratified or matched on, propensity score within multivariable models because (1) there were too few events within some propensity score strata to perform stratification and (2) unmatched saxagliptin initiators would have been excluded, reducing power to detect associations.
Meta-analyses of each outcome across data sources were performed as data permitted. The presence of heterogeneity in HRs across data sources was evaluated using the I2 statistic.17
We performed sensitivity analyses to (1) evaluate outcomes when the cohorts were expanded to include patients prescribed/dispensed insulin or GLP-1 agonists and (2) examine the effect of unmeasured confounders on HRs of each outcome associated with saxagliptin use.18 Details appear in online supplementary methods. Data were analyzed using SAS V.9.4.
Results
Patient characteristics
We identified 110 740 eligible saxagliptin initiators and 913 384 eligible other OAD initiators (see online supplementary figure S1a-d). These patients’ characteristics are presented in tables 1 (UK) and 2 (USA). Across the four data sources, the average follow-up ranged from 6.8 to 8.1 months among saxagliptin initiators and 5.6 to 7.0 months among other OAD initiators. Saxagliptin initiators more commonly had hypertension and were more frequently prescribed/dispensed antihyperlipidemics, antihypertensives, and prior OAD therapy (metformin, sulfonylureas, and/or thiazolidinediones). Metformin initiators constituted the majority of the comparator OAD cohort.
The numbers of confirmed events following medical record review and PPVs of diagnostic coding algorithms varied by end point and data source (see online supplementary table S12).
Risk of MACE
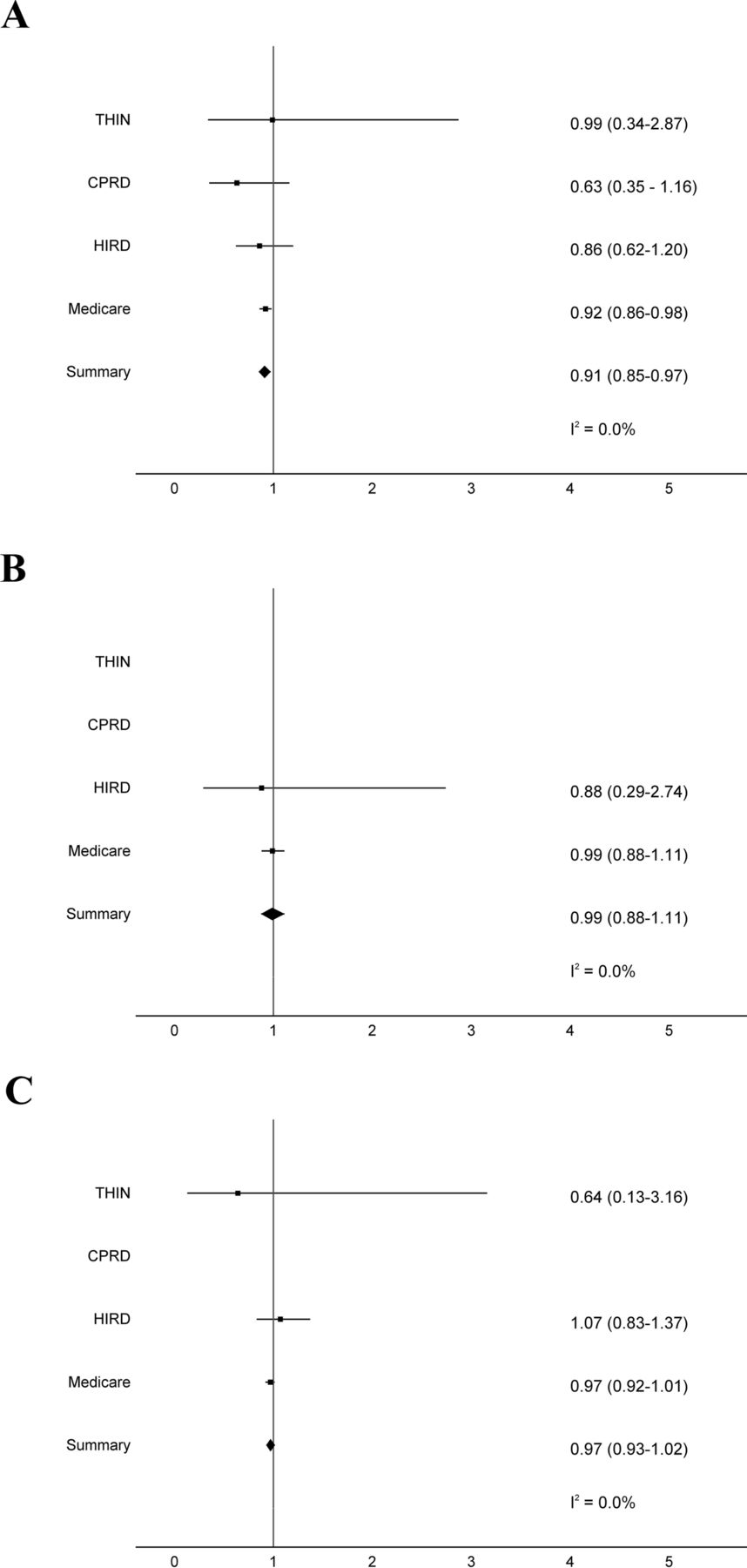
There was no increased risk of MACE associated with saxagliptin initiation within any of the four data sources. Within Medicare, the incidence rate and risk of MACE was lower for saxagliptin than for other OAD initiators (HR 0.92, 95% CI, 0.86 to 0.98; table 3). Meta-analysis of results across the four data sources demonstrated a lower risk of MACE associated with saxagliptin initiation (HR 0.91, 95% CI 0.85 to 0.97; figure 1A).
{kind=link}
Meta-analyses of HRs (with 95% CIs) of hospitalization with and/or death due to a major adverse cardiovascular event (A), hospitalization for acute kidney injury (B), and hospitalization for infection (C) across the data sources. CPRD, Clinical Practice Research Datalink; HIRD, HealthCore Integrated Research Database; THIN, The Health Improvement Network.
Incidence rates and HRs of outcomes, by data source
Risk of ALF
There was no increased risk of ALF associated with saxagliptin initiation within each data source (table 3). Across the data sources, no saxagliptin initiators and only one comparator OAD initiator (within Medicare) developed ALF, and medical record review determined this was not drug-related. There were too few ALF events to permit meta-analysis.
Risk of AKI
There was no association between saxagliptin initiation and hospitalization for AKI across the data sources (table 3). Meta-analysis across the USA data sources demonstrated no increased risk of AKI with saxagliptin (HR 0.99, 95% CI, 0.88 to 1.11; figure 1B). The UK data sources were not included in the meta-analysis due to few events.
Risk of infection
Across the data sources, there was no association between saxagliptin initiation and hospitalization for infection (table 3). Meta-analysis of results within THIN, Medicare, and HIRD showed no increased risk of this outcome (HR 0.97, 95% CI 0.93 to 1.02; figure 1C). Data from CPRD were not included because the diagnostic coding algorithm had <80% PPV.
Risk of severe hypersensitivity reactions
There were no significant differences in incidence rates or risk of hospitalization for severe hypersensitivity reactions between saxagliptin and other OAD initiators within each data source (table 3). Across the data sources, only one saxagliptin initiator (within Medicare) had an event confirmed by medical record review. Due to the low number of events, we were unable to perform a meta-analysis.
Sensitivity analyses
When the cohorts were expanded to include patients who were prescribed/dispensed insulin and/or a GLP-1 agonist, there was no increased risk of any outcome within each data source (see online supplementary table S13). Across all data sources, sensitivity analyses to determine the potential impact of unmeasured confounders determined that the HRs for each outcome were not sensitive to unmeasured confounding (see onlinesupplementary table S14).
Conclusions
This very large family of cohort studies of two UK healthcare record data sources and two USA claims-based data sources (analyzed individually and combined via meta-analyses) found no increases in incidence rates or risk of hospitalization with or death due to MACE, hospitalization with ALF, or hospitalization for AKI, infection, or severe hypersensitivity reaction among new initiators of saxagliptin compared with new initiators of OADs in non-DPP-4 inhibitor classes.
Consistent with previously published studies,19–25 we found no evidence of increased risk of MACE associated with saxagliptin initiation. A meta-analysis of 53 clinical trials for DPP-4 inhibitors found a reduced risk of MACE among patients prescribed DPP-4 inhibitors compared with those prescribed placebo or comparator therapies.19 Similarly, the Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus–Thrombolysis in Myocardial Infarction 53 trial found that saxagliptin use was not associated with a higher risk of AMI, ischemic stroke, or cardiovascular death.23 Our findings provide further evidence that saxagliptin is not associated with an increased risk of MACE in practice settings.
Post hoc analyses of clinical trials data have suggested no increased risk of acute liver injury with saxagliptin.25 We observed no association between saxagliptin initiation and hospitalization with ALF. Notably, across the four data sources, we observed no ALF events among saxagliptin initiators and only one event among comparator OAD initiators.
Consistent with prior clinical trials,26 27 this study demonstrated no association between saxagliptin initiation and hospitalization for AKI, confirming the renal safety of saxagliptin in real-world settings.
Our finding of no association between saxagliptin initiation and infection is consistent with the overall low incidence rates and risk of infection among DPP-4 inhibitors observed in clinical trials.25 28 A pooled analysis of 20 randomized trials of saxagliptin, prescribed as a monotherapy or add-on therapy, found similar incidence rates of infection between saxagliptin users (24.2 per 100 patient-years) and the control groups (21.7 per 100 patient-years).25 A meta-analysis of 30 randomized trials comparing vildagliptin with placebo also found no increased risk of infections.28 However, one meta-analysis of randomized trials of DPP-4 inhibitors (sitagliptin, vildagliptin, saxagliptin) observed an increased risk of nasopharyngitis (risk ratio 1.2, 95% CI 1.0 to 1.4) and urinary tract infections (risk ratio 1.5, 95% CI 1.0 to 2.2) associated with DPP-4 inhibitor use.29
Our findings demonstrating no increased risk of hospitalizations for severe hypersensitivity reactions among saxagliptin initiators are in contrast with prior clinical trials analyses.25 30 In one pooled analysis, urticaria and facial edema occurred more commonly among patients with type 2 diabetes mellitus who initiated saxagliptin compared with those receiving placebo (incidence 1.5% vs 0.4%).30 A separate analysis of 20 clinical trials comparing hypersensitivity reactions between users of saxagliptin and comparator drugs or placebo found that rates of these events were more common among saxagliptin initiators (incidence rate ratio 1.67, 95% CI, 1.01 to 2.87); however, incidence rates for both groups were low (saxagliptin, 1.3 per 100 person-years; control, 0.8 per 100 person-years).25 Differences in the definitions of hypersensitivity events between those studies and ours likely accounts for these disparate findings.
Our study has several potential limitations. There is the possibility for unmeasured confounding since not all clinically important variables are consistently captured across these data sources. However, we performed sensitivity analyses to determine the effect of unmeasured variables on measures of effect within each data source and observed that the results were insensitive to unmeasured confounding. Misclassification of both the exposure and outcome is possible. Misclassification of new initiators of saxagliptin or comparator OADs could exist if providers supplied samples or drug rebate cards for varying durations to patients, with no record in the data sources. Moreover, misclassification of new initiators may occur in the UK data sources if the patient was initially prescribed the OAD by a specialist as the general practitioner may not have a record of this initial prescription. Finally, some analyses were based on coding algorithms with <80% PPV or with very few events, limiting our assessment of risk for these end points. However, very few events in a population exceeding 1.0 million indicate that any risk must be very small.
Our analyses have a number of strengths. Using four data sources, rather than a single database, provided a larger sample for safety analyses and allowed inclusion of patients across a variety of settings, within both the USA and UK, and from private and public health insurance plans, enhancing generalizability. We controlled for numerous potential confounding variables using propensity scores. We used standardized definitions for end points and evaluated the validity of diagnostic coding algorithms for these events using medical records. Finally, the 95% CIs surrounding the relative hazards were generally very narrow, indicating a high level of power. Although we cannot completely rule out associations with the outcomes, we can eliminate the likelihood of moderate-to-large associations.
In conclusion, saxagliptin initiation was not associated with increased rates of hospitalized MACE, ALF, AKI, infections, or severe hypersensitivity reactions. The low risk of these events among saxagliptin initiators, particularly in such large study populations, provides real-world evidence of the safety of this medication.
Acknowledgments
The authors thank Jennifer Wood, PhD, MPH of Bristol-Myers Squibb and Eileen Ming, MPH, ScD of Epi Excellence for their critical review of the manuscript.
References
Footnotes
Contributors VLR and BLS developed the study concept and design. DMC, KH, SEK, PPR, DJM, AJA, KRR, HB, AMG and DBE participated in the acquisition of data. CWN, QL, QW and JAR performed statistical analyses. VLR, DMC, MES, CWN, SEK, PPR and BLS conducted interpretation of the data. VLR and DMC drafted the manuscript. All authors provided critical revisions of the manuscript. VLR is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding This study was funded by AstraZeneca and the study’s sponsors approved the protocol and had the right to provide non-binding comments on this manuscript, but were excluded from all analyses involving Medicare data.
Competing interests VLR, DMC, MES, CWN, JAR, QL, QW, SC, KH, SEK, PPR, DJM, AJA, KRR and BLS received funding from AstraZeneca through their employers. AMG and HB are employees of CPRD and THIN, respectively. KH and DBE are employees of HealthCore. SEK has consulted for Pfizer, Merck and Bayer, all unrelated to this manuscript.
Ethics approval This study was approved by the UK Independent Scientific Advisory Committees for CPRD (Protocol 10_149RMn) and THIN (Protocol 11-039V), Quorum Review Institutional Review Board for HIRD, and Institutional Review Boards of the University of Pennsylvania and Rutgers University. A data use agreement was obtained from the Centers for Medicare and Medicaid Services.
Provenance and peer review Not commissioned; externally peer reviewed.