Article Text

Abstract
Objective DS-8500a is a novel G protein-coupled receptor 119 agonist being developed for the treatment of type 2 diabetes. The study objective was to assess the efficacy and safety of DS-8500a in Japanese patients with type 2 diabetes.
Research design and methods In this double-blind, parallel-group, phase II study, 99 Japanese patients with type 2 diabetes were randomized to receive placebo, or DS-8500a 10 mg or 75 mg once daily for 28 days. The primary efficacy endpoint was change in the 24-hour weighted mean glucose (WMG) from baseline (day −1) to day 28. Other endpoints included changes in fasting plasma glucose, postprandial glucose, lipids, and safety.
Results The 24-hour WMG decreased significantly after 28 days of treatment in the 10 mg and 75 mg groups with placebo-subtracted least squares mean differences (95% CI) of −0.74 (−1.29 to –0.19) mmol/L and −1.05 (−1.59 to –0.50) mmol/L, respectively. Reductions in 24-hour WMG in both DS-8500a groups were observed on day 14 and were greater on day 28 than on day 14. The reductions in fasting plasma glucose and 2-hour postprandial glucose were significantly greater in the 75 mg DS-8500a group versus placebo. Total cholesterol, low-density lipoprotein cholesterol, and triglycerides decreased significantly; high-density lipoprotein cholesterol increased significantly in the 75 mg group versus placebo. Both doses of DS-8500a were well tolerated without significant treatment-related adverse events, hypoglycemia, or discontinuations due to adverse events.
Conclusions DS-8500a significantly improved glycemic control and lipids and was well tolerated over 28 days of administration in Japanese patients with type 2 diabetes.
Trial registration number NCT02222350; Post-results.
- G protein-coupled receptor 119 agonist
- glycemic control
- Japanese
- phase II clinical trial
- type 2 diabetes
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- G protein-coupled receptor 119 agonist
- glycemic control
- Japanese
- phase II clinical trial
- type 2 diabetes
Significance of this study
What is already known about this subject?
Agonists targeting G protein-coupled receptor (GPR) 119 represent one of the newest classes of oral antidiabetic drugs in clinical development.
Several agonists of GPR119 have been developed; however, the results of clinical trials have been disappointing because the drugs showed a marked waning of effect after 14–28 days.
What are the new findings?
Unlike other GPR119 agonists, we observed no waning in the glucose-lowering effects of DS-8500a in terms of the reductions in 24-hour weighted mean glucose, fasting plasma glucose, and postprandial glucose.
DS-8500a was well tolerated without serious adverse events, hypoglycemia, or discontinuations due to adverse events.
How might these results change the focus of research or clinical practice?
Our results provide critical support for larger and longer phase III clinical studies of DS-8500a, which has potential as an oral antidiabetic agent for the care of patients with type 2 diabetes.
Introduction
Multiple treatment options for type 2 diabetes are available. The Japan Diabetes Society1 recommends administering oral hypoglycemic agents, insulin, or glucagon-like peptide-1 (GLP-1) receptor agonists as initial therapy based on patient preference, the clinical subset of diabetes of an individual patient, and drug characteristics, with the goal of reducing blood glucose levels while minimizing side effects, especially hypoglycemia, if the HbA1c target is not achieved despite lifestyle modifications. The American Diabetes Association2 guidelines suggest metformin as a first-line treatment together with lifestyle modifications. Physicians should consider adding a second oral agent, a GLP-1 receptor agonist, or insulin if initial therapy is unable to achieve glycemic control targets. Most antidiabetic drugs improve glycemic control by reducing systemic glucose concentrations (and hence HbA1c) through increased tissue glucose uptake (by stimulating endogenous insulin secretion or providing exogenous insulin), increased peripheral glucose disposal, inhibition of renal glucose reabsorption, or decreased intestinal glucose absorption. If we consider that pancreatic β cell dysfunction is a major contributor to the progression of type 2 diabetes, novel drugs that improve insulin secretion from pancreatic β cell via a mechanism distinct from currently available drugs may provide important advances to the treatment of this disease.
G protein-coupled receptor 119 (GPR119) is a newly identified receptor that is predominantly expressed on intestinal L cells and pancreatic β cells. In vitro and animal studies have helped to elucidate the physiologic roles of GPR119. Intestinal GPR119 contributes to regulating glycemic control by enhancing the secretion of GLP-1 and glucose-dependent insulinotropic peptide.3 Activation of GPR119 in β cells also enhances glucose-dependent insulin secretion.4 Moreover, it was reported that GPR119 stimulates proglucagon gene expression in enteroendocrine cells5 and enhances β cell replication and islet graft function.6
Based on these physiologic roles, GPR119 has been considered as a novel treatment target for type 2 diabetes,7 prompting the development of GPR119 agonists. Unfortunately, the development of several GPR119 agonists was terminated after the initial clinical studies because of weak glucose-lowering effects or lack of efficacy after repeated dosing of up to 14 days.8 9
DS-8500a is a novel, orally available GPR119 agonist being developed for the treatment of type 2 diabetes. Several preclinical studies have been performed to confirm the pharmacologic effects of DS-8500a. DS-8500a increased intracellular cyclic adenosine monophosphate in human GPR119-expressing CHO-K1 cells in a concentration-dependent manner with a half maximal effective concentration (EC50) of 51.5 nmol/L and had no significant effect on multiple receptors, channels, or transporters studied (66 molecular targets, half maximal inhibitory concentration (IC50) >10 μmol/L).10 In animal studies, DS-8500a enhanced glucose-stimulated insulin secretion in Sprague-Dawley rats and promoted GLP-1 secretion in Zucker fatty rats.10 In addition, DS-8500a preserved β cell function and prevented increases in glycohemoglobin concentrations in rodent models of type 2 diabetes.11
In healthy Japanese male volunteers, DS-8500a was well tolerated at single and repeated doses of up to 100 mg for 7 days.12 Pharmacokinetic/pharmacodynamic modeling of preclinical and clinical results after repeated dosing suggested that doses of 10–75 mg once daily will have pharmacodynamic effects in patients with diabetes.
Based on the results of preclinical pharmacology and clinical pharmacokinetics studies, we conducted a 28-day study to examine the efficacy and safety of a low and high dose of DS-8500a in Japanese patients with type 2 diabetes.
Materials and methods
Ethical considerations
This study was performed in accordance with the principles of Good Clinical Practice and the ethical standards for human experimentation established by the Declaration of Helsinki. The study was approved by the institutional review boards at all participating sites. The clinical trial registration number was JapicCTI-142597, NCT02222350 (ClinicalTrials.gov).
Study population
Adults aged 20–69 years with type 2 diabetes were eligible if (1) they were not using oral hypoglycemic agents and were on diet and exercise therapy, or (2) they were on monotherapy with an oral antidiabetic drug (sulfonylurea, glinides, metformin, α-glucosidase inhibitors, dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium–glucose cotransporter-2 inhibitors, or GLP-1 analogs), provided that the drug could be temporarily discontinued during the study. Other eligibility criteria were HbA1c of ≥6.5% to <9.5% (≥48 mmol/mol to <80 mmol/mol) at the screening visit (day −42) and ≥7.0% to <10.0% at the start of the run-in period (day −14) for patients previously using antidiabetic drugs, or HbA1c of ≥7.0% to <10.0% (≥53 mmol/mol to <86 mmol/mol) at the start of the run-in period (day −14) for treatment-naïve patients, and body mass index of ≥18.5 kg/m2 to <35.0 kg/m2.
The major exclusion criteria were age ≥70 years; history of type 1 diabetes or diabetic ketoacidosis; fasting plasma glucose (FPG) ≥13.3 mmol/L; history of clinically significant diabetic retinopathy, diabetic nephropathy, or diabetic neuropathy; currently on or indicated for insulin therapy; use of a thiazolidinedione within 90 days before screening; poorly controlled blood pressure; liver disease; renal impairment; acute coronary syndrome, stroke, coronary or peripheral artery revascularization within 6 months of screening; or current diagnosis of clinically serious diseases; pregnant and breast feeding; and patients deemed unsuitable for participation by the investigators for any other reason.
All patients provided written informed consent.
Study design and treatments
This randomized, double-blind, placebo-controlled, parallel-group, phase II study was conducted at three clinical trial centers in Japan (Heishinkai OCROM Clinic, Osaka; PS Clinic, Fukuoka; and Sumida Hospital, Tokyo).
The study comprised four periods: a 28-day washout period for patients previously using an antidiabetic drug; a 14-day single-blind, placebo run-in period for all patients; a 28-day randomized, placebo-controlled treatment period; and a 7-day post-treatment follow-up period. All patients were hospitalized from day −2 to day 2, from day 13 to day 15, and from day 27 to day 29 for 24-hour glycemic assessments under standardized caloric intake conditions. Randomization was performed on day 1.
Patients were randomized into three groups, placebo, 10 mg DS-8500a, and 75 mg DS-8500a, in a 1:1:1 ratio with stratification by HbA1c (≤8.0 %/>8.0%; ≤64 mmol/mol/>64 mmol/mol) via an interactive web response system. DS-8500a and matching placebo tablets were administered in a double-blind manner for 28 days.
Pharmacokinetic/pharmacodynamic modeling from preclinical pharmacology studies and from the clinical PK profiles after repeated dosing suggests that doses of 10–75 mg once daily will likely achieve pharmacodynamic effects in patients with diabetes. The 75 mg dose of DS-8500a was selected from a safety perspective, after reviewing the results of a prior 7-day multiple-dose study in Japanese individuals12 and a preclinical study in cynomolgus monkeys.13 The lower dose of 10 mg was chosen to provide preliminary dose–response data.
After obtaining informed consent, diet and exercise therapy was to continue unchanged throughout the study period.
During the three in-hospitalization periods, patients received standardized meals that provided a total of 1600 kcal per day, in three meals (500 kcal for breakfast and lunch, and 600 kcal for dinner). Each meal comprised 60% carbohydrates, 25% fat, and 15% protein.
Patients were informed about the risk of hypoglycemia, including symptoms and actions to take in the event of symptoms. Patients were instructed to contact the study site in the event of hypoglycemic symptoms.
Study endpoints and measures
The primary endpoint was the change in 24-hour weighted mean glucose (WMG) from baseline to day 28. Secondary efficacy endpoints included the change in 24-hour WMG from baseline to day 14, and the changes in FPG, 2-hour postprandial plasma glucose (PPG), plasma glucose, serum insulin, C-peptide, active GLP-1, and peptide YY from baseline to day 28.
In each hospitalization period, 15 blood samples were obtained within the 24-hour measurement period, as follows: the day before administration; immediately before administration (0 hour); and at 0.5, 1, 2, 4, 4.5, 5, 6, 8, 9, 9.5, 10, 11, 13, and 24 hours after administration. The glucose concentrations were used to calculate the 24-hour WMG as the plasma glucose area under the glucose concentration–time curve from 0 to 24 hours divided by 24, in accordance with a previous study.14
Treatment-emergent adverse events (TEAEs), drug-related TEAEs, vital signs, and clinical and laboratory tests were monitored throughout the study. TEAEs were assessed in terms of their seriousness, outcome, severity, and causality to the study drug. TEAEs were coded in accordance with the Medical Dictionary for Regulatory Activities (MedDRA/J) V.17.0 by system organ class and preferred term.
Sampling time points
The sampling time points relative to meals were as follows: The 2-hour PPG samples were obtained 2 hours after breakfast, lunch, and dinner. FPG was measured at least 10 hours after the last meal. Plasma glucose was measured on days −1 and 28 (before breakfast and 0.5, 1, 2, and 4 hours after breakfast; 0.5, 1, 2, and 4 hours after lunch; before dinner and 0.5, 1, 2, and 4 hours after dinner); and on day 1 and the day after treatment completion (24 hours after breakfast the day before). Serum insulin was measured on days −1 and 28 (before breakfast; 0.5, 1, 2, and 4 hours after breakfast; 0.5, 1, 2, and 4 hours after lunch; before dinner; 0.5, 1, 2, and 4 hours after dinner); and on day 1 and the day after treatment completion (24 hours after breakfast the day before). C-peptide was measured on days −1 and 28 (before breakfast; 0.5, 1, 2, and 4 hours after breakfast). Active GLP-1 and peptide YY were measured on days −1 and 28 (before breakfast; 0.5, 1, 2, and 4 hours after breakfast). HbA1c was measured on days 1 and 28 (before breakfast). Glycoalbumin was measured on days 1 and 28 (before breakfast).
Active GLP-1 immunoassay with ethanol and solid-phase extractions
Active GLP-1 was measured using the Glucagon-Like Peptide-1 (Active) ELISA kit (catalog no EGLP-35K; Millipore, Billerica, Massachusetts, USA), which recognizes GLP-1 isoforms at the following cross-reactivities: GLP-1 (1–37), 0.2%; GLP-1 (7–37), 99.5%; GLP-1 (9–37), not detected; GLP-1 (1–36) amide, 0.2%; GLP-1 (7–36) amide, 100%; and GLP-1 (9–36) amide, not detected.15
Statistical analyses
An effect size (the mean intergroup difference divided by the common SD) of 0.9 was assumed for the primary endpoint of change in 24-hour WMG from baseline to day 28 between one of the DS-8500a groups and the placebo group. Therefore, a sample size of 30 per group would ensure a 90% statistical power at a two-sided significance level of 5%, with a dropout rate of approximately 10%.
Data analyses were conducted in the full analysis set (FAS), per protocol set (PPS), and safety analysis set, as appropriate. The FAS comprised all randomized patients who satisfied the protocol-specified inclusion criteria, who received at least one dose of the allocated study drug, and whose 24-hour WMG was determined predose and at least once postdose. The PPS comprised all patients in the FAS after excluding those who met any of the exclusion criteria or had a serious protocol deviation. The safety analysis set comprised all randomized patients treated with at least one dose of the study drug in the treatment period.
The baseline characteristics are summarized descriptively using the mean, SD, and the number and frequency of patients, as appropriate, and were conducted in the FAS. The primary endpoint, the change in 24-hour WMG, was analyzed by analysis of covariance (ANCOVA), which included the treatment group as a fixed effect and baseline 24-hour WMG as a covariate. The primary endpoint was assessed in the FAS and the analysis was repeated in the PPS as a sensitivity analysis. Secondary endpoints were analyzed using ANCOVA with treatment group as a fixed effect and the baseline values as a covariate.
Missing data were not imputed for the analysis of the primary endpoint, although sensitivity analyses were conducted after considering missing data using the last observation carried forward method.
Statistical analyses were performed using SAS V.9.2 system release.
Results
Study population
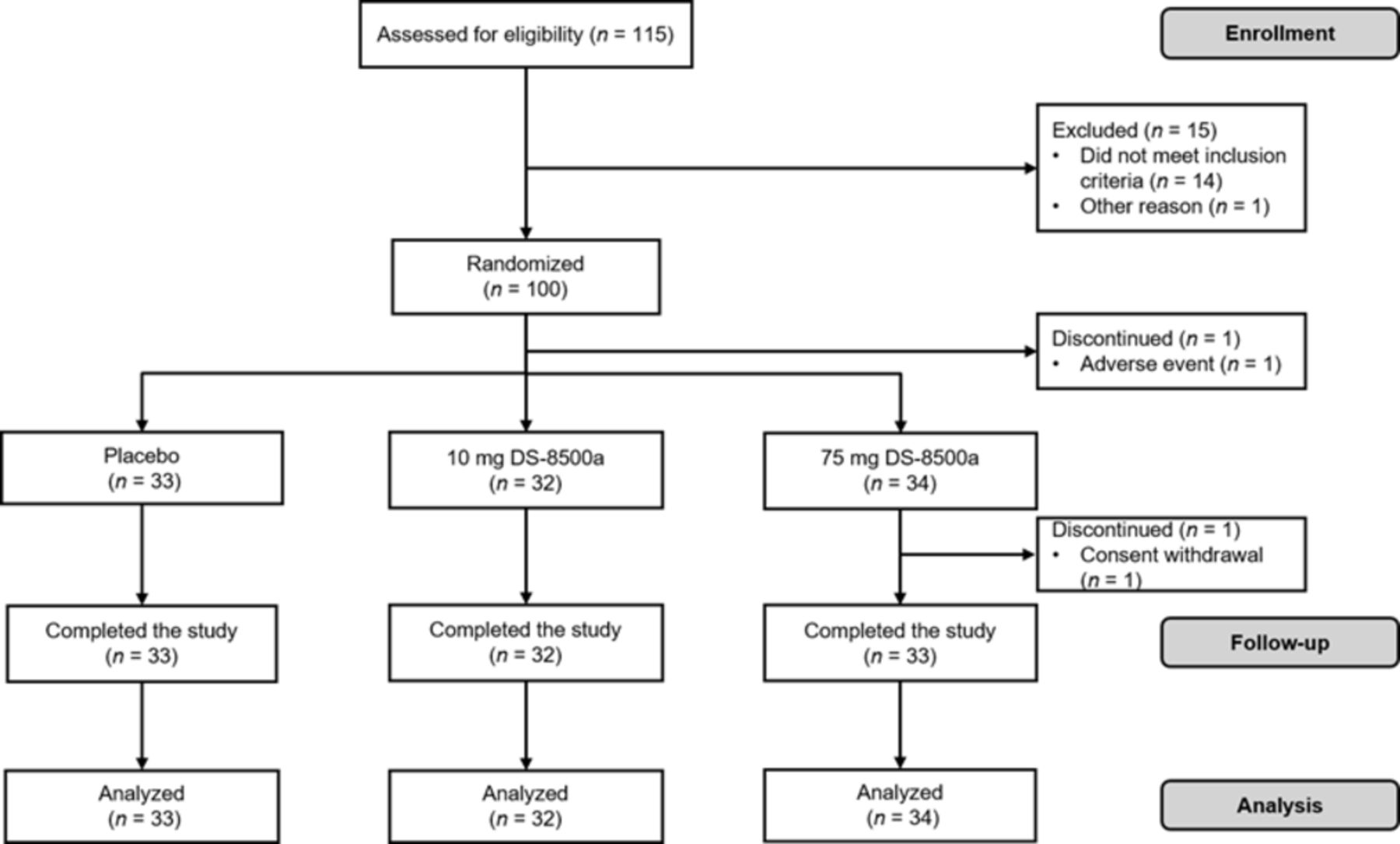
The study was performed between 12 July 2014 and 11 November 2014. A total of 115 patients with type 2 diabetes were screened, 100 eligible patients were randomized, and one patient randomized to the 75 mg group discontinued before administration of the study drug (figure 1). Therefore, 33, 32, and 34 patients were randomized to the placebo, 10 mg DS-8500a, and 75 mg DS-8500a groups, respectively, and completed all study assessments. These 99 patients were included in both the FAS and PPS analyses. The study included 88 men (88.9%). The mean diabetes duration, HbA1c, and FPG were 6.89 years, 8.01% (64 mmol/mol), and 8.79 mmol/L, respectively. Sixteen patients (16.2%) had previously used an oral antidiabetic drug and 11 (11.1%) were using a lipid-lowering drug. The baseline demographic and glycemic characteristics of the three groups were generally similar (table 1).

Patient characteristics at the screening visit
Changes in WMG
Between baseline and day 28 of treatment, the 24-hour WMG decreased from 11.59 to 11.03 mmol/L in the 10 mg group, decreased from 11.26 to 10.42 mmol/L in the 75 mg group, and increased from 11.08 to 11.34 mmol/L in the placebo group. DS-8500a significantly reduced 24-hour WMG compared with placebo on both days 14 and 28 (figure 2). The least square (LS) mean between-treatment differences (95% CI) from baseline to day 28 were −0.74 mmol/L (−1.29 to –0.19 mmol/L; p=0.0093) and −1.05 mmol/L (−1.59 to –0.50 mmol/L; p=0.0002) in the 10 mg and 75 mg groups, respectively. The responses observed in the 10 mg and 75 mg DS-8500a groups were greater on day 28 than on day 14. Similar results were shown for the primary endpoint in the PPS. Similar results were also shown after considering missing data using the last observation carried forward method.

{kind=link}
{kind=link}
Change in 24-h WMG. Values are shown as the least squares mean change±SE, with p values versus placebo. WMG, weighted mean glucose.
Glycemic profiles and insulin section
Table 2 shows the changes in other glycemic parameters from baseline to day 28. DS-8500a at 75 mg was associated with significant reductions in FPG (p<0.01) and 2-hour PPG after breakfast (p<0.01), lunch (p<0.01), and dinner (p<0.01) as compared with placebo. DS-8500a at 10 mg significantly reduced 2-hour PPG after lunch (p<0.05) and dinner (p<0.05) compared with placebo. Although reductions in 2-hour PPG after breakfast and FPG were observed in the 10 mg group between baseline and day 28, these changes were not statistically significant relative to the changes in the placebo group. Twenty-four-hour plasma glucose excursion profiles are shown in online supplementary figure S1. Markers of β cell function and insulin secretion included the areas under the concentration–time curves from 0 to 4 hours (AUC0–4h) for glucose, C-peptide, and insulin, as well as the ratios insulin AUC0–4h/glucose AUC0–4h and C-peptide AUC0–4h/glucose AUC0–4h. As expected, the glucose AUC0–4h was significantly reduced in both DS-8500a groups relative to placebo (both p<0.05). In addition, C-peptide AUC0–4h (p<0.05), insulin AUC0–4h/glucose AUC0–4h (p<0.01) and C-peptide AUC0–4h/glucose AUC0–4h (p<0.05) were significantly increased in the 75 mg group compared with placebo, but not in the 10 mg group. Twenty-four-hour serum insulin profiles at baseline and on day 28 are shown in online supplementary figure S2. Glycoalbumin was measured in the placebo group as well as both dose treatment groups at baseline and on day 28. There was a significant decrease in mean glycoalbumin levels from baseline to day 28 in the 75 mg group (p<0.0001); however, no significant changes from baseline were seen in the 10 mg group or placebo group (data not shown).
Supplementary file 1
![[SP1.jpg]](https://drc.bmj.com/content/bmjdrc/5/1/e000424/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
Supplementary file 2
![[SP2.jpg]](https://drc.bmj.com/content/bmjdrc/5/1/e000424/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
Effects of DS-8500a on glycemic and lipid parameters
Lipid profiles
Improvements in lipid profiles were observed after treatment with DS-8500a for 28 days. In the 75 mg group, the LS mean changes (adjusted for placebo) from baseline to day 28 in total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and triglyceride concentrations were −0.417, –0.225, and −0.732 mmol/L, respectively (p<0.01, p<0.05, and p<0.01, respectively), corresponding to changes of 8.4%, 7.0%, and 35.9%, respectively. The LS mean change in high-density lipoprotein cholesterol (HDL-C) was +0.166 mmol/L (p<0.001), corresponding to a change of 15.9% (table 2). Triglyceride concentrations on day 28 were also significantly lower in the 10 mg DS-8500a group compared with placebo. The changes in lipid concentrations were not correlated with the changes in 24-hour WMG (data not shown).
Other pharmacodynamics parameters
There were no apparent differences in active GLP-1 or peptide YY (PYY) measured predose or at 0.5, 1, or 2 hours postdose on day 14 or 28 among the 10 mg and 75 mg DS-8500a, and placebo groups (data not shown).
Safety
The incidence of TEAEs in each group is shown in table 3. Two, four, and three patients in the placebo, 10 mg, and 75 mg groups, respectively, reported TEAEs. In the DS-8500a groups, TEAEs were acute pharyngitis, oropharyngeal pain, gingivitis, hordeolum, triglyceride increased, and urinary occult blood positive. All of the TEAEs resolved, none were considered serious or severe, and no patients discontinued because of a TEAE. There were no drug-related TEAEs or episodes of hypoglycemia/hypoglycemia symptoms, and there were no apparent safety signals in terms of vital signs, laboratory tests, 12-lead electrocardiography, funduscopy, or physical examination. There were no apparent changes in body weight, blood pressure, or pulse rate during the study in any group.
Adverse events, vital signs, and laboratory variables
Discussion
This study showed that administration of DS-8500a at a dose of both 10 mg and 75 mg once daily for 28 days was associated with significant improvements in glycemic and lipid variables compared with placebo in Japanese patients with type 2 diabetes. The effects on glycemic variables persisted on both day 14 and day 28, and were seen for both fasting and postprandial parameters. The magnitude of improvements was smaller in the 10 mg DS-8500a group than in the 75 mg DS-8500a group. We also observed improvements in lipid concentrations and estimates of β cell function, especially in the 75 mg group, but there were no apparent changes in GLP-1 and PYY concentrations in either group. Both doses of DS-8500a were safe and well tolerated, and there were no episodes of hypoglycemia. Furthermore, neither dose of DS-8500a was associated with changes in body weight, blood pressure, or pulse rate within the context of this study.
Some aspects of the present results warrant particular mention. First, we observed significant increases in the C-peptide concentration, C-peptide AUC0–4h, insulin AUC0–4h/glucose AUC0–4h, and C-peptide AUC0–4h/glucose AUC0–4h, especially in the 75 mg DS-8500a group. These variables may relate to β cell function and insulin secretory capacity in response to glucose, and the finding is consistent with the hypothesis that DS-8500a, a GPR119 agonist, enhances glucose-dependent insulin secretion and may improve β cell function. However, we found no apparent changes in GLP-1 or PYY concentrations in any group, despite prior animal studies showing that GPR119 is involved in regulating the secretion of GLP-13 and that DS-8500a enhanced GLP-1 secretion in rats.10 This discrepancy between pharmacologic and clinical results may be related to differences in the species studied, or to differences in the chemical structures8 9 of the molecules being studied.
Another finding of interest is that no hypoglycemia was reported as a TEAE in this study. GPR119 was reported to enhance glucose-dependent insulin secretion, as well as GLP-1 and glucose-dependent insulinotropic peptide.3 4 Accordingly, we speculate that GPR119 agonists might avoid hypoglycemia by promoting insulin secretion in the presence of elevated glucose concentrations, and its effects might be attenuated once glucose concentrations decline. We conducted a hyperglycemic clamp study to evaluate the insulin secretory capacity of DS-8500a and found that it enhanced the insulin secretory capacity (first-phase and second-phase insulin secretion) in patients with type 2 diabetes over the 4-week period.16 However, this would need to be verified in future studies.
Several pharmacokinetic, pharmacodynamic, and phase II studies have been conducted for other GPR119 agonists, including GSK1292263,9 MBX-2982,17 PSN821,18 LEZ763,19 and JNJ-38431055,8 20 but the results of these studies were inconsistent. For example, Nunez et al9 reported that GSK1292263 did not improve glycemic control in patients with type 2 diabetes, but it did markedly increase circulating PYY concentrations, and the gut hormone effects of GSK1292263 were modulated by coadministration of metformin or sitagliptin. Similarly, Katz et al8 reported that JNJ-38431055 had limited glucose-lowering effects in patients with type 2 diabetes. By contrast, PSN821 was reported to significantly lower glucose concentrations, and influenced cardiometabolic factors in patients with type 2 diabetes.18 Meanwhile, MBX-2982 was reported in an abstract to have significantly reduced glucose excursions and glucagon concentrations in mixed meal tolerance tests in men with impaired fasting glucose, and enhanced glucose-dependent insulin secretion in graded glucose infusion tests,17 although the full results are not yet available.
In these other studies, the glucose-lowering effects of these investigational drugs appeared to wane or deteriorate after about 14 days of administration relative to their effects within 1 day of administration,8 9 which might explain their limited effects on glycemic control. By contrast, DS-8500a elicited progressive reductions in glucose concentrations from 14 to 28 days. Some evidence of tachyphylaxis was observed in studies of other GRP119 agonists,8 9 17 but not in patients treated with either dose of DS-8500a in the present study. Although the reason for the differences in efficacy of GPR119 agonists is unclear, it is conceivable that the chemical structures of each agonist might help explain the differences. For example, some GPR119 agonists contain a piperidine ring as fatty acid derivatives. The piperidine ring may undergo conformational changes, and the administered drug may be present in two conformations, with one conformation acting as an agonist and the other as an antagonist for GPR119.21 DS-8500a lacks a piperidine ring, which might explain the observed efficacy in the present study.
Both doses of DS-8500a significantly reduced 24-hour WMG in our study. In a study of sitagliptin in Japanese patients with type 2 diabetes,14 50 mg twice daily or 100 mg once daily has been shown to reduce 24-hour WMG compared with placebo. Although there are differences in the patients’ characteristics, study design and conduct between the two studies that might limit direct comparisons, the results nonetheless suggest that the improvements in glycemic control in our study are of clinical significance and may be comparable to those achieved with other established antidiabetic drugs.
The present study also revealed significant reductions in TC, LDL-C, and triglycerides, and a significant increase in HDL-C in the 75 mg DS-8500a group compared with placebo. Improvements in lipid profiles were also reported for other GPR119 agonists, including GSK129226322 and PSN821.18 In the present study, the changes in 24-hour WMG and lipids were not correlated, which suggests that these changes might be mediated via different mechanisms of action. Preliminary data suggest that GPR119 agonists slow the systemic appearance of cholesterol after an oral cholesterol load in rats, or that they might promote the clearance of chylomicrons or other triglyceride-rich lipoproteins from the circulation.23 Additional studies are clearly needed to evaluate the mechanism of action of GPR119 agonists on lipids.
Limitations of this study include its relatively short duration, small sample size, limited number of doses tested, a higher proportion of men than women, and that the study included only Japanese patients with diabetes. Interestingly, type 2 diabetes mellitus in Asians is characterized by β cell dysfunction rather than insulin resistance. In contrast, type 2 diabetes mellitus in non-Asians is characterized by insulin-resistance.24 25 Insulin secretagogues, such as GLP-1 agonists and DPP-4 inhibitors, have a greater efficacy in Asian patients with diabetes compared with non-Asians. Therefore, DS-8500a (a GPR119 agonist) might also be more effective for Asian patients with diabetes because it is an insulin secretagogue.
In conclusion, DS-8500a was effective in improving glycemic and lipid profiles, and was safe and well tolerated in Japanese patients with type 2 diabetes. The results of this study indicate that a GPR119 agonist can be clinically effective in patients with type 2 diabetes. Further clinical studies with a larger size and longer duration in which DS-8500a is administered alone or in combination with other agents, such as metformin or a DPP-4 inhibitor, are needed to confirm the results of the current monotherapy study.
References
Footnotes
Contributors NI, HSC, ST, TW, KS, YN, and TT contributed to study design, data analysis, and writing of the manuscript. As corresponding author, TT is guarantor for the content of this article.
Funding Medical writing support was provided by Nicholas D Smith, PhD (Edanz Medical Writing) and was funded by Daiichi Sankyo.
Competing interests NI has acted as a consultant for Novo Nordisk and Arkray Marketing; has received research support from Kissei Pharmaceutical, Eli Lilly Japan KK, Novartis Pharma KK, Daiichi Sankyo, Sanofi KK, Pfizer Japan, Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical Company, Japan Tobacco, Kyowa Hakko Kirin, Dainippon Sumitomo Pharma, Astellas Pharma, Merck Sharp & Dohme, Sanwa Kagaku Kenkyusho, Boehringer Ingelheim Japan, Ono Pharmaceutical, Tsumura & Co, Fujifilm Pharma, Shiratori Pharmaceutical, and Taisho Toyama Pharmaceutical; has served on the speaker’s bureau for Merck Sharp & Dohme, Astellas Pharma, Eisai, Sanofi KK, Novo Nordisk, Kowa Pharmaceutical, Ono Pharmaceutical, Dainippon Sumitomo Pharma, Daiichi Sankyo, Eli Lilly Japan KK, Boehringer Ingelheim Japan, Arkray Marketing, Japan Tobacco, Takeda Pharmaceutical Company, and Johnson & Johnson; and has acted as the medical adviser on a clinical trial performed by Daiichi-Sankyo. HSC is an employee of Daiichi Sankyo Pharma Development. ST is an employee of Daiichi Sankyo Development. TW, KS, YN, and TT are employees of Daiichi Sankyo.
Ethics approval IRB.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional information is available.