Article Text

Abstract
Objective Obesity-associated metabolic dysfunction increases the risk of multiple diseases such as type 2 diabetes and cardiovascular disease. The importance of the co-stimulatory CD40-CD40L dyad in diet-induced obesity (DIO), with opposing phenotypes arising when either the receptor (aggravating) or the ligand (protective) is deleted, has been described previously. The functions of CD40 and CD40L are cell type dependent. As co-stimulation via T cell-mediated CD40L is essential for driving inflammation, we here investigate the role of T cell CD40L in DIO.
Research design and methods CD4CreCD40Lfl/fl mice on a C57BL/6 background were generated and subjected to DIO by administration of 15 weeks of high fat diet (HFD).
Results HFD-fed CD4CreCD40Lfl/fl mice had similar weight gain, adipocyte sizes, plasma cholesterol and triglyceride levels as their wild-type (WT) counterparts. Insulin and glucose tolerance were comparable, although CD4CreCD40Lfl/fl mice did have a decreased plasma insulin concentration, suggesting a minor improvement of insulin resistance. Furthermore, although the degree of hepatosteatosis was similar in both genotypes, the gene expression of fatty acid synthase 1 and ATP-citrate lyase had decreased, whereas expression of peroxisome proliferator-activated receptor-α had increased in livers of CD4CreCD40Lfl/fl mice, suggesting decreased hepatic lipid uptake in absence of T cell CD40L.
Moreover, CD4CreCD40Lfl/fl mice displayed significantly lower numbers of effector memory CD4+ T cells and regulatory T cells in blood and lymphoid organs compared with WT. However, immune cell composition and inflammatory status of the adipose tissue was similar in CD4CreCD40Lfl/fl and WT mice.
Conclusions T cell CD40L deficiency results in a minor improvement of insulin sensitivity and hepatic steatosis in DIO, despite the strong decrease in effector T cells and regulatory T cells in blood and lymphoid organs. Our data indicate that other CD40L-expressing cell types are more relevant in the pathogenesis of obesity-associated metabolic dysfunction.
- type 2 diabetes
- obesity
- T cells
- T lymphocyte activation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
The CD40L-CD40 co-stimulatory dyad orchestrates the deleterious low-grade inflammation found in diet-induced obesity (DIO).
Full body deletion of the CD40L co-stimulatory molecule has protective effects in the pathogenesis of obesity and its associated metabolic dysfunction in mice.
What are the new findings?
Our study found that T cell-specific deletion of CD40L does not emulate the full body deletion of CD40L, as we found only minor improvement of obesity and its associated metabolic dysfunction in the T cell CD40L deficient mice.
T cell CD40L deficient mice have decreased CD4+ effector T cells and regulatory T cells; however, this does not have an impact on the low-grade inflammation associated with obesity.
How might these results change the focus of research or clinical practice?
T cell CD40L impacts T cell differentiation, however, shifts in pro-inflammatory and anti-inflammatory T cell subsets may negate each other, eliminating the effect a deletion of either subset may have in different tissues during DIO.
The results presented in this manuscript indicate that targeting CD40L for therapeutics should be refined to target a specific cell type, other than T cell CD40L, to have a significant impact in DIO and its associated metabolic dysfunction.
Introduction
Global obesity rates have tripled since 1975, with 39% of the population worldwide classified as obese, defined by a body mass index (BMI) ≥25 kg/m2, in 2016.1 Obesity and its associated metabolic dysregulation, characterized by hyperglycemia, insulin resistance and chronic low-grade (adipose tissue) inflammation, has been shown to increase the risk of multiple diseases including type 2 diabetes (T2D), cardiovascular disease, cancer, non-alcoholic fatty liver disease, and non-alcoholic steatohepatitis.2–5
During obesity, an excess of nutrients, especially fatty acids, cause adipocyte expansion. This leads to adipocyte hypoxia and oxidative stress, which ultimately results in adipocyte dysfunction and apoptosis.3 4 6 Adipocytes respond to this stress by secretion of pro-inflammatory cytokines and adipokines such as CCL2 (MCP-1), interferon (IFN)-γ, interleukin (IL)6, and leptin, thereby recruiting and activating immune cells to the adipose tissue.3 4 In obese adipose tissue, almost the entire spectrum of immune cells including macrophages, eosinophils, T cells, and B cells have been proven to play major roles in adipose tissue dysfunctional metabolism and inflammation.3 7 8
Co-stimulatory molecules are master regulators of immune responses and as such play an important role in experimental models of obesity and in human disease.9 A co-stimulatory dyad with protective as well as deleterious effects in the pathogenesis of obesity and its associated metabolic dysfunction is CD40L-CD40, which are members of the tumor necrosis factor (TNF) and TNF receptor (TNFR) superfamily, respectively.9–12 CD40 is expressed on antigen-presenting cells (APCs), including B cells, dendritic cells and macrophages, as well as on endothelial cells (ECs) and adipocytes. The ligand CD40L is mainly found on CD4+ T cells, platelets, ECs, vascular smooth muscle cells (VSMCs), and is present in plasma in its soluble form (sCD40L).9–12
Mice with a genetic deficiency of CD40 have worsened progression of diet-induced obesity (DIO). This is evidenced by an earlier onset of insulin resistance, increased adipose tissue inflammation and liver steatosis,13–16 indicating that CD40 is protective in obesity-mediated metabolic dysfunction.
In contrast, CD40L has deleterious effects. Stimulation of adipocytes with sCD40L induced inflammatory cytokine expression.13 Furthermore, data from human cohort studies have found a positive correlation between sCD40L plasma levels, insulin resistance, and hypercholesterolemia in obese subjects.17–22 Accordingly, CD40L deficient mice with DIO exhibited reduced adipose tissue inflammation, accompanied by an increase in adipose tissue regulatory T cells (Treg), reduced insulin resistance, and a decrease in hepatic steatosis.14 19 Another study showed that CD40L deficiency resulted in reduced macrophage infiltration into adipose tissue and a reduction of IgG antibodies against oxidized lipids, but failed to show an effect on insulin resistance or hepatic steatosis.23
The complexity of the co-stimulatory CD40-CD40L dyad in obesity is emphasised by the opposing phenotypes that arise in mice deficient for either protein. As CD40L is expressed on multiple cell types, including T cells, platelets, and ECs that all exert different effector functions during obesity-associated metabolic dysfunction, it is important to determine the role of these different CD40L-containing cell types in DIO.14 23–25 Here, we elucidate the role of T cell CD40L, a strong driver of T cell-mediated immune responses, in DIO using T cell CD40L deficient mice.
Methods
Animals
CD40Lfl/fl mice were generated by introducing LoxP sites upstream and downstream of exon 3 (custom design, Ozgene, Bentley, Australia). CD40Lfl/fl mice were crossed to CD4-Cre transgenic mice (Stock No 017336,26 27 Jackson Laboratory, Bar Harbor, Maine, USA) to generate CD4CreCD40Lfl/fl mice. Littermates not containing the CD4-Cre transgene were used as controls (CD40Lfl/fl, in the following referred to as WT). All mice were bred and maintained at the animal facility of the Amsterdam UMC (location AMC), Amsterdam.
Study design
Male CD4CreCD40Lfl/fl mice (n=15) and CD40Lfl/fl littermates (n=14) had ad libitum access to water and high fat diet (HFD; SNIFF-D12492, energy 22% from carbohydrates, 24% from protein and 54% from fat, Sniff, Soest, Germany), or a standard fat diet (n=8 per group) (SFD; SNIFF-D12450B, energy 65% from carbohydrates, 26% from protein, and 9% from fat) for 15 weeks starting at 7 weeks of age. Body weight was monitored weekly. At the end of the study, animals were euthanized using a combination of ketamine (100 mg/kg) and xylazine (30 mg/kg) injected intraperitoneally for the collection of blood and organs (spleen, lymph nodes (LN), epididymal adipose tissue (EpAT), and subcutaneous adipose tissue (ScAT)).
Glucose and insulin tolerance tests
Glucose and insulin tolerance tests (GTT/ITT) were performed 12 weeks after introduction of the diet. For the GTT, 5.5 hours fasted mice were injected intraperitoneally with glucose (1 mg/g body weight, Sigma-Aldrich Zwijndrecht, The Netherlands). For the ITT, mice were fasted 4 hours and injected intraperitoneally with insulin (0.75 mU/g body weight, Sigma-Aldrich). Glucose levels were measured in whole blood from the tail using a glucometer (Bayercontour, Basel, Switzerland) at 0, 15, 30, 45, 75, and 120 min in the GTT and at 0, 15, 30, 60, 90, and 120 min in the ITT.
Histology
EpAT and liver were collected and fixed in 4% paraformaldehyde and embedded in paraffin. EpAT sections (4 µm) were stained with H&E or stained using immunohistochemistry with CD45 (BD Biosciences, San Jose, California, USA), MAC3 (BD Biosciences) and CD3 (AbD serotec, Kidlington, UK) antibodies. Adipocyte size was determined on H&E stained tissue using ImageJ. H&E sections (4 µm) of liver were scored for the degree of liver inflammation and steatosis by a pathologist (MJJG), who was blinded for the experimental conditions. Grades were scored (0–5), 0=absent, 1=minimal, 2=mild, 3=moderate, 4=high, 5=severe. Livers were embedded in Tissue Tek OCT (Sakura Finetek, Alphen aan den Rijn, The Netherlands) and frozen at −80°C. These livers were sectioned into 8 µm sections for staining with Oil Red O (Sigma-Aldrich). All histological analyses were done by an observer blinded for the experimental conditions.
Cholesterol and triglyceride measurements
Plasma total cholesterol and triglyceride concentrations were measured using standard enzymatic methods (CHOD-PAP and GPO-PAP; Roche Diagnostics, Almere, The Netherlands).
Flow cytometry
For flow cytometric analysis, blood was obtained by cardiac puncture with an EDTA-coated syringe. Spleen and LNs were removed and collected in phosphate-buffered saline (PBS). EpAT and ScAT were prepared separately by mincing into small pieces and digested using liberase (0.25 mg/mL Dulbecco’s Modified Eagle Medium, Roche) for 45 min at 37°C. Spleen, LN, and digested adipose tissue samples were passed through a 70 µm nylon mesh (BD Biosciences). The stromal vascular fraction (SVF) was collected by pelleting of the suspension and diluted in fluorescence-activated cell sorting (FACS) buffer (0.5% bovine serum albumin, 0.01% NaN3 and 2 mM EDTA in PBS). Blood and spleen were incubated with hypotonic lysis buffer (8.4 g NH4Cl and 0.84 g NaHCO3 in 1 L miliQ) to remove erythrocytes. Cell suspensions were blocked with CD16/32 antibody (eBioscience, Waltham, Massachusetts, USA) in FACS buffer to prevent non-specific binding. Indicated tissues were incubated with CD45, CD19, CD62L, CD44, FoxP3, Ly6C (eBioscience), CD3, CD8, CD38, CXCR3, CD40L (Biolegend, San Diego, California, USA), CD4, CD11b, Ly6G, SiglecF (BD Biosciences), and F4/80 (AbD serotec) antibodies. Fluorescence was measured by flow cytometry (FACSCanto II, BD Biosciences) and analysed with FlowJo software V.10 (Tree star).
In vitro stimulation of lymphocytes
LNs were removed from CD4CreCD40Lfl/fl mice (n=3) and CD40Lfl/fl littermates (n=3) for isolation of lymphocytes. LNs were passed through a 70 µm nylon mesh (BD Biosciences) and whole LN cell suspension was treated with CD3/CD28 stimulating Dynabeads (ThermoFisher Scientific, Waltham) in RPMI (Gibco, Waltham) with 5% FCS, for 24 hours in 37°C with 5% CO2. T cells were subsequently analysed by flow cytometry for expression of CD40L.
Gene expression analysis
Lymphocytes were sorted by BD FACSAria II to obtain CD4+ and CD8+ populations and lysed using a RNeasy Kit (Qiagen, Hilden, Germany). Total RNA of EpAT and liver was extracted using Trizol (Invitrogen, Waltham). All RNA was reverse transcribed to cDNA using an iScript cDNA synthesis kit (Bio-Rad, Lunteren, The Netherlands). Quantitative PCR was performed with a SYBR green PCR kit (Applied Biosystems, Waltham) on a ViiA7 real-time PCR system (Applied Biosystems). Analysis was performed using the housekeeping genes β-actin and GAPDH.
Plasma cytokine measurements
Plasma cytokine levels were measured on a Luminex 200 system using an 18-plex mouse Procartaplex panel (eBioscience) containing markers for IFN-γ, TNF-α, IL1β, IL4, IL6, IL10, IL12p70, IL17A, IL17F, IL23, IP-10, G-CSF, GM-CSF, MCP-1, MCP-3, MIP-1α, MIP-1β, and MIP-2. The Olink Mouse Exploratory panel (Olink Proteomics, Uppsala, Sweden) of 92 high-quality assays for murine proteins was used for identification of plasma proteins related to disease in addition to inflammatory markers. The assay was performed via the Olink Proteomics services (https://www.olink.com/analysis-service/).
Statistical analysis
Results are presented as mean±SD for n=8 mice per group for SFD and n=14 or 15 mice per group for HFD, unless stated otherwise in the figure legend. Prism 7.03 software (GraphPad) was used for statistical analysis. Data were analysed by unpaired t-test, Mann-Whitney U test or two-way analysis of variance with Sidaks multiple correction test, depending on normality determined by the D’Agostino-Pearson omnibus normality test. P values <0.05 were considered to be statistically significant.
Results
T cell CD40L deficiency does not affect weight gain or insulin resistance during DIO
CD4CreCD40Lfl/fl mice were successfully generated and had a~78% depletion of CD40L in CD3+CD4+ T cells (online supplementary figure S1). When consuming an HFD, body weight gain significantly increased compared with mice fed an SFD. However, deficiency of T cell CD40L did not affect total weight gain in mice fed either diet (figure 1A). Although liver weights were equal between both genotypes (figure 1D), EpAT weight was slightly reduced in CD4CreCD40Lfl/fl mice after HFD feeding (p=0.068, figure 1B). In accordance, we also observed a slight but non-significant (p=0.31) decrease in adipocyte size in EpAT of CD4CreCD40Lfl/fl mice fed an HFD (figure 1C). Furthermore, we observed slight decreases in plasma cholesterol (~12%, p=0.19) and plasma triglycerides (~20%, p=0.19) in HFD-fed CD4CreCD40Lfl/fl mice (figure 1E,F).
Supplemental material
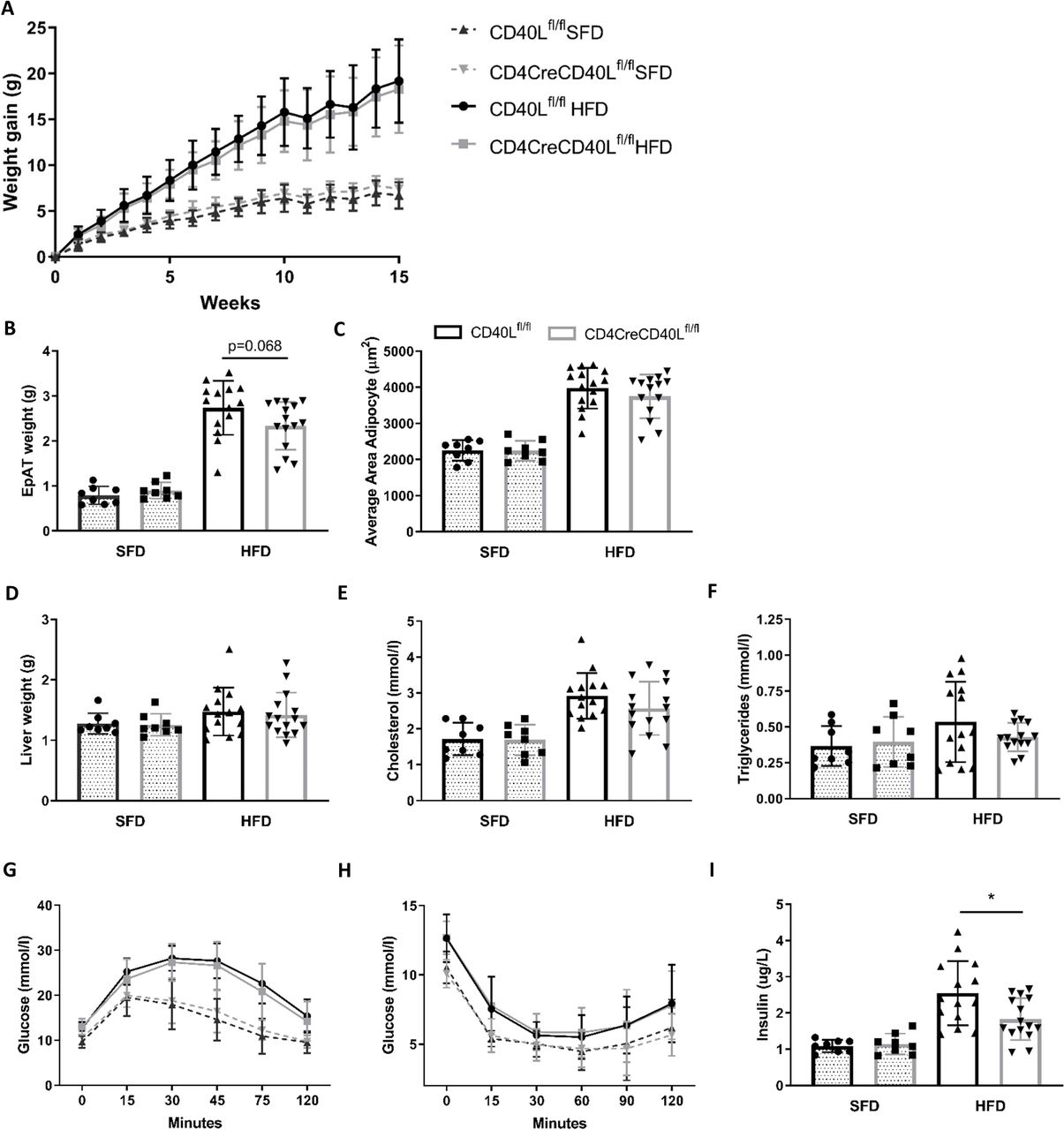
Weight gain, glucose and insulin tolerance tests show no differences between WT and T cell CD40L deficient mice. (A) Body weight gain of HFD (n=14 and 15/group) and SFD (n=8/group) of CD4CreCD40Lfl/fl and WT mice during 15 weeks of diet. (B) Epididymal adipose tissue weight (EpAT) after 15 weeks of diet. (C) Average adipocyte size in area (µm2) for HFD and SFD. (D) Liver tissue weight after 15 weeks of diet. (E, F) Plasma cholesterol and triglyceride concentrations in mmol/L. (G) Glucose tolerance tests HFD (n=15/group) and SFD (n=8/group) at week 12 of dietary intervention. (H) Insulin tolerance tests HFD (n=15/group) and SFD (n=8/group) at week 12 of dietary intervention. (I) Insulin concentration in plasma of mice fasted for 5 hours. Data are presented as mean±SD, *p<0.05. HFD, high fat diet; SFD, standard fat diet; WT, wild type.
Mice fed an HFD for 12 weeks had significantly worsened glucose tolerance and insulin sensitivity compared with SFD (figure 1G,H). However, no differences were observed at the individual time points of either the GTT or ITT, between CD4CreCD40Lfl/fl and WT mice when fed SFD or HFD. Interestingly, we did observe a significant decrease in fasted plasma insulin levels in CD4CreCD40Lfl/fl mice (figure 1I). This indicates that CD4CreCD40Lfl/fl mice do have slightly improved insulin sensitivity, as reduced hyperinsulinemia is associated with metabolically healthier and more insulin sensitive tissue.28 Transcription analysis of genes involved in glucose regulation, fatty acid transport, fatty acid synthesis and lipolysis in EpAT was performed to confirm these findings. Concordant with the improvement in insulin sensitivity, GLUT4, a gene involved in glucose regulation and a mediator of cellular insulin sensitivity, had increased expression in EpAT of CD4CreCD40Lfl/fl mice (p=0.053, online supplemntary figure S2A). Other genes such as scavenger receptor CD36 and fatty acid synthase 1 (FAS1), genes involved in lipid metabolism, exhibited a similar expression profile in both genotypes (online supplementary figure S2A).
CD4CreCD40Lfl/fl mice exhibit slightly reduced hepatosteatosis
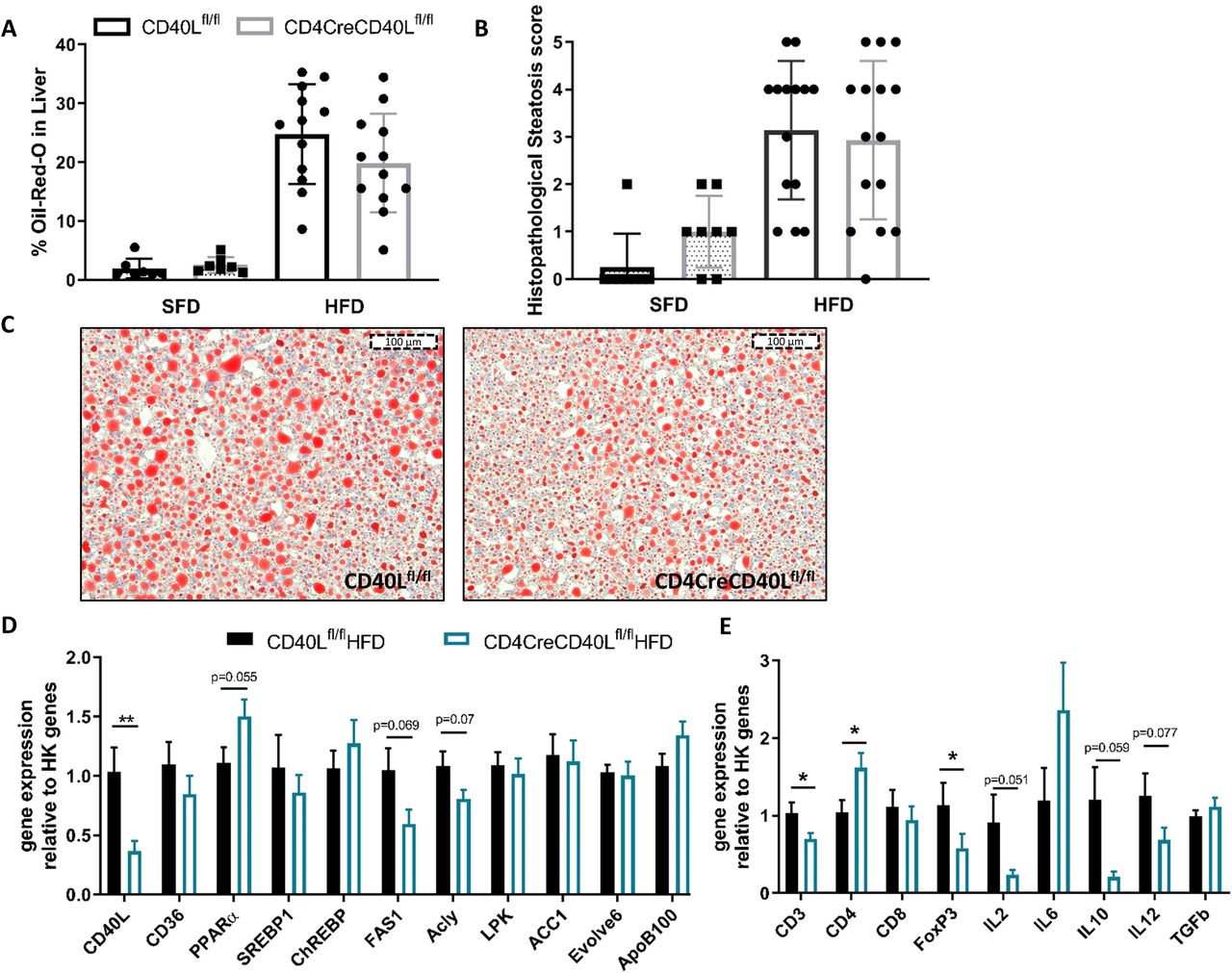
To investigate the effect of T cell CD40L deficiency in DIO on liver pathology, the degree of steatosis and inflammation was determined. Histological analysis revealed that deficiency of T cell CD40L did not affect the degree of steatosis and bile duct proliferation in mice fed either SFD or HFD (figure 2A, online supplementary figure S3). The amount of steatosis, measured by determining the Oil-red-O positive area, showed a non-significant but ~20% decrease in the CD4CreCD40Lfl/fl mice (24.8% in WT vs 19.9% in CD4CreCD40Lfl/fl, p=0.166, figure 2B,C). These findings are reflected by the liver metabolism gene transcription analysis, as CD4CreCD40Lfl/fl mice have decreased expression of genes involved in de novo lipid synthesis such as FAS1 and ATP-citrate lyase (Acly) (figure 2D). Furthermore, the expression of transcription factor peroxisome proliferator-activated receptor alpha (PPARα), which promotes fatty acid uptake and lipogenesis was increased in livers of CD4CreCD40Lfl/fl mice (figure 2D), indicating a decreased hepatic lipid uptake in absence of T cell CD40L.
Supplemental material

CD40L deficiency has a minor ameliorating effect on hepatosteatosis and hepatic inflammation. (A) Quantification of Oil-red-O (ORO) staining in CD4CreCD40Lfl/fl and WT mice of HFD and SFD. (B) Histopathological score of liver steatosis, as scored by a pathologist (0=absent, 1=minimal, 2=mild, 3=moderate, 4=high, 5=severe). (C) Histological representative images of ORO in liver. (D) mRNA expression of liver lipid metabolism genes relative to housekeeping (HK) genes. (E) mRNA expression of inflammatory genes in the liver relative to HK genes. Data (A–C) is presented as mean±SD, (D/E) HFD (n=14 and 15/group) and SFD (n=8/group) are presented as mean±SEM, *p<0.05, **p<0.01. HFD, high fat diet; SFD, standard fat diet; WT, wild type.
Histological analysis of hepatic inflammation showed a low overall degree of inflammation in both genotypes (online supplementary figure S3). However, livers of CD4CreCD40Lfl/fl mice displayed reduced expression of pro-inflammatory cytokines IL2 and IL12 (figure 2E). These data indicate that mice deficient in T cell CD40L are slightly protected against obesity-associated hepatic steatosis and inflammation.
Supplemental material
Deficiency of T cell CD40L impairs T cell activation but fails to reduce diet-induced adipose tissue inflammation
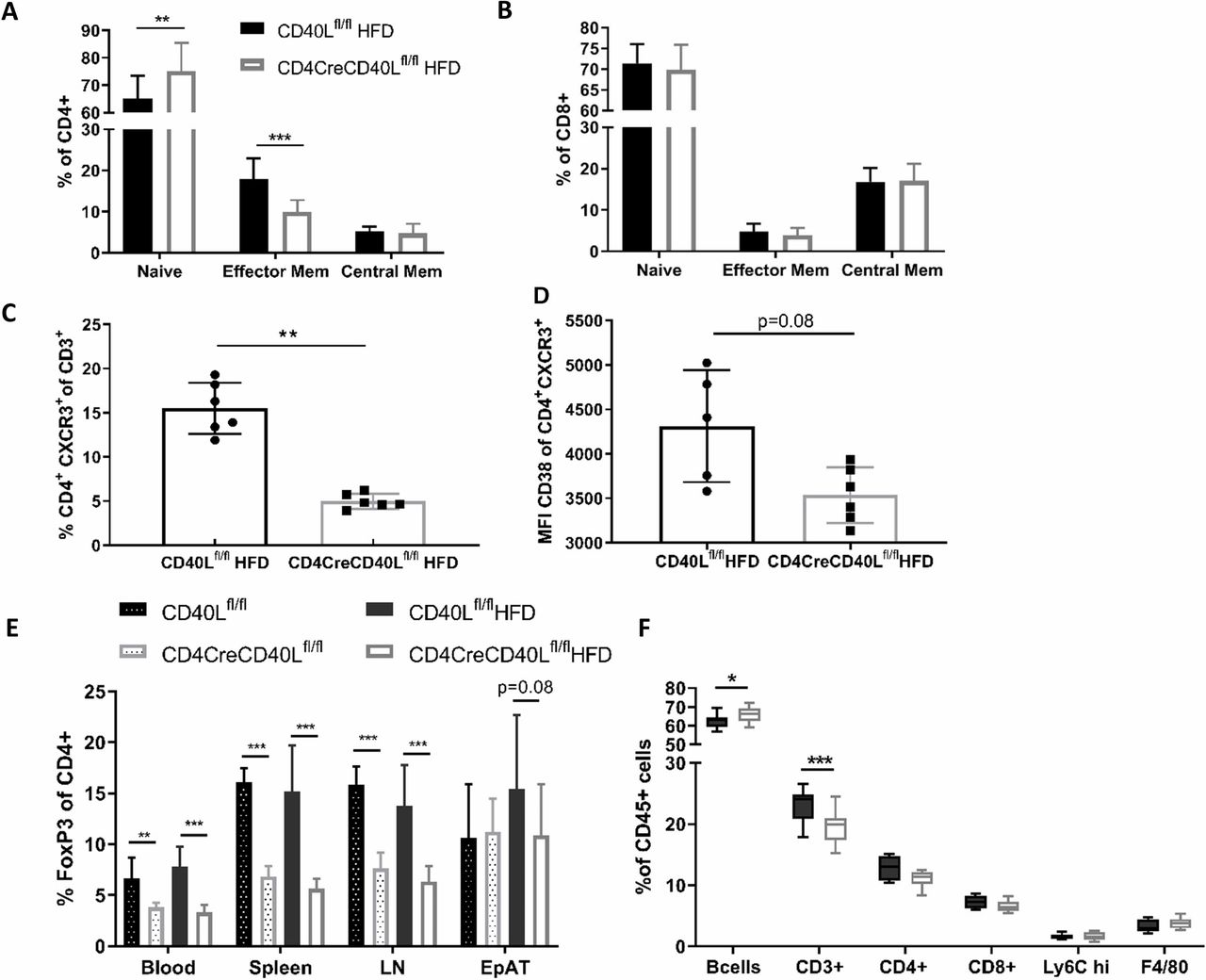
As CD40L is crucial in T cell activation, T cell activation status was analysed by flow cytometry. In blood and lymphoid organs, deficiency of T cell CD40L resulted in a reduction of CD4+CD62L-CD44+ effector memory cells in both the HFD-fed and SFD-fed mice (figure 3A, online supplementary figure S4). The fraction of CD4+CD62L+CD44+ central memory cells remained similar between the groups and consequently the fraction of CD4+CD62L+CD44− naïve cells had increased (figure 3A). The CD8+ naïve and effector subsets were not affected by deficiency of T cell CD40L (figure 3B). Expression of other T cell effector markers, such as chemokine receptor CXCR3 and cyclic ADP ribose hydrolase glycoprotein CD38, were reduced on CD4+ T cells in CD4CreCD40Lfl/fl mice (figure 3C,D). Furthermore, cells that did express CXCR3 had reduced CD38 expression, implicating that these cells were transitioning into a central memory phenotype (figure 3C,D). Combined, these results are in line with the increase in naïve T cells as well as the apparent prompt transition from effector T cells into memory T cells.
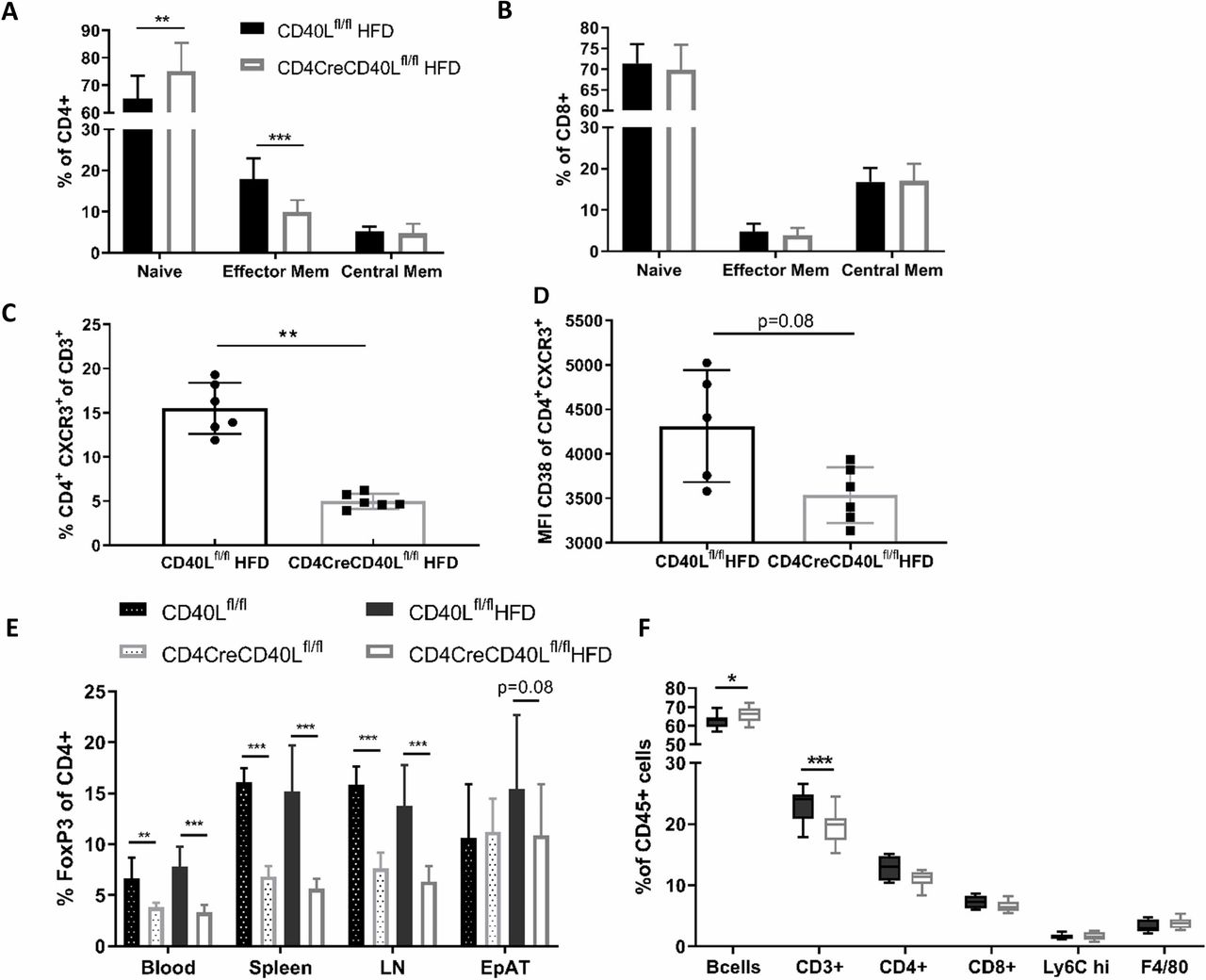
Flow cytometric analysis of T cell subtypes reveals a more naïve phenotype in CD40L deficient mice. (A) Distribution of CD4+ T cell subsets in spleen; CD62L+ CD44− naïve cells, CD62L+ CD44+ central memory cells and CD62L− CD44+ effector memory cells of HFD mice. (B) Distribution of CD8+ T cell subsets in spleen; CD62L+ CD44- naïve cells, CD62L+ CD44+ central memory cells, and CD62L− CD44+ effector memory cells. (C, D) Flow cytometric analysis of CD4+ effector cells and the expression of CXCR3 and CD38 in spleen (n=6/group). (E) Flow cytometric analysis of CD3+FoxP3+ Tregs in different organs of HFD and SFD mice. (F) Flow cytometric analysis of CD45+ subsets in spleen. Data are represented HFD (n=14 and 15/group) and SFD (n=8/group) with mean±SD. *P<0.05, **p<0.01, ***p<0.001. EpAT, epididymal adipose tissue; HFD, high fat diet; LN, lymph node; SFD, standard fat diet; Treg, regulatory T cell.
Furthermore, the fraction of FoxP3+ Tregs in both the SFD and HFD groups was decreased in blood and lymphoid organs of CD4CreCD40Lfl/fl mice (p<0.001, figure 3E). This, together with a profound reduction in IL2 expression (online supplementary figure S1D), is in line with the decreased T cell activation that is necessary for natural Treg formation.29
Flow cytometric analysis of cells of the innate immune system revealed that these were unaffected by the T cell-specific CD40L deficiency. No significant differences between the genotypes were observed in neutrophils, eosinophils, macrophages, and monocytes, irrespective of their activation status (MHCII+) or origin (blood and lymphoid organs) (figure 3F, online supplementary figure S4).
To further determine the effect of T cell CD40L deficiency on systemic inflammation, plasma cytokine levels were analysed. Both genotypes on SFD and HFD showed baseline low concentrations of cytokines and chemokines. However, slight differences could be observed in both pro-inflammatory and anti-inflammatory cytokines, but major shifts that may attribute to functional significance in inflammation between CD4CreCD40Lfl/fl and WT mice could not be observed (online supplementary figure S5A,B). This was furthermore illustrated by the Olink Proteomics analysis of 92 plasma proteins for broad disease interaction markers. Multiple comparison statistical analysis of HFD mice plasma revealed that the data did not cluster on genotype, indicating that the variation is mostly genotype independent for these plasma proteins during DIO (online supplementary figure S6).
Supplemental material
Adipose tissue immune cell infiltration is not affected by T cell CD40L deficiency
Although CD4CreCD40Lflfl mice exhibit a major decrease in effector T cells in blood and lymphoid organs, adipose tissue inflammation was unaffected (figure 4A–C). Flow cytometry revealed no relevant differences in CD45+ lymphocyte populations (CD3+, CD4+, CD8+, FoxP3+) or innate cell populations of monocytes, macrophages, eosinophils or neutrophils infiltrating the EpAT (figure 4B), similar data were found in ScAT (data not shown). This was confirmed by histology, where the number of infiltrated CD45+ leukocytes, Mac3+ macrophages, and CD3+ T cells increased on HFD feeding, but did not differ between CD4CreCD40Lflfl and WT mice (figure 4C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Epididymal adipose tissue (EpAT) histology shows no differences in immune cell infiltration. (A) Histological representative images of CD45+ staining in EpAT. (B) Flow cytometry analysis of CD45+ subsets in EpAT. (C) Quantification of CD45+, MAC3+ macrophages or CD3+ lymphocytes per 100 adipocytes. (D) mRNA expression of cellular markers and inflammatory genes in EpAT relative to HK genes. Data (B/C) are represented as HFD (n=14 and 15/group) and SFD (n=8/group) with mean±SD, (D) is presented as HFD (n=14 and 15/group) and SFD (n=8/group) with mean±SEM. HFD, high fat diet; SFD, standard fat diet.
Furthermore, transcript analysis of whole EpAT showed similar expression levels of pro-inflammatory genes including CD40L, IFN-γ, IL2, IL6, IL12, and TNF (figure 4D, online supplementary figure S2A), indicating that other CD40L expressing cells such as ECs, VSMCs, and platelets redundantly express CD40L, and that the overall inflammation in the adipose tissue seems to be similar between the T cell CD40L deficient and WT mice.
Discussion
Despite the fact that CD40L has been shown to play a pivotal role in the pathogenesis of DIO, our findings indicate that T cell-specific CD40L is not a main driver of obesity-induced metabolic dysfunction. Previous studies report that total-body CD40L deficient mice subjected to DIO exhibit a strong decrease in adipose tissue inflammation, increased insulin sensitivity, and decreased hepatosteatosis.14 19 23 However, our data reveal that T cell CD40L deficiency only results in minor improvements in insulin sensitivity and hepatic inflammation. These results indicate that other CD40L expressing cell types, such as platelets or ECs, may play a more prominent role in obesity-associated adipose tissue inflammation and metabolic dysfunction than T cell CD40L.
Indeed, there are indications that other CD40L expressing cells play an important role in DIO. Recently, it was reported that ECs experience increased oxidative stress, as observed in db/db mice, as a consequence of increased CD40L expression during obesity.19 Additionally, EC oxidative stress caused by activated CD40L expressing platelets can enhance expression of endothelial-CD40L, while expression of soluble and cell surface chemoattractant CCL2 (MCP-1) was also increased.30 In accordance, data obtained from obese full-body CD40L deficient mice showed that adipose tissue exhibited decreased expression of CCL2, which was associated with a decrease in immune cell infiltration of the adipose tissue.14 19 23 As our study found no differences in CCL2 expression and adipose tissue inflammation, other CD40L expressing cells such as ECs, VSMCs, and platelets may mediate CD40L-driven immune cell infiltration in adipose tissue.
The role of platelet CD40L in DIO is emphasized by several clinical studies correlating sCD40L with insulin resistance. Subjects with a BMI >30 kg/m2 have increased plasma levels of sCD40L, which is mainly platelet derived.17–21 Increased plasma sCD40L is correlates with fasting glucose, insulin resistance and leukocyte counts in human cross-sectional studies.17–20 Furthermore, when patients with T2D were compared with age-matched controls, plasma sCD40L was significantly higher, independent of cholesterol, waist circumference, BMI or sex.17 This suggests that platelet CD40L may play a more important role in metabolic dysfunction than T cell CD40L. Combined, these data show the crucial roles of induction and maintenance of inflammation in DIO by CD40L expressing cells other than T cells.
In our mouse model, we found that 78% of T cell CD40L was depleted, which resulted in major changes in T cell subsets and their expression profiles. Although we cannot rule out residual T cell CD40L activity, the fact that T effector cell numbers have decreased substantially suggests that the remaining CD40L+ T cell population plays a minor role. Our results show that T cell CD40L is pivotal for the generation of CD4+ effector T cells and CD4+ Tregs, cell types that are considered major regulators of obesity-induced adipose tissue inflammation and insulin resistance.31 32 Adipose tissue T cell quantity and T cell subtype content correlates with BMI and waist circumference in patients with obesity and T2D. During obesity, the number of Th2 cells and anti-inflammatory Tregs decreases in adipose tissue and are replaced with a gross increase in pro-inflammatory Th1 cells.31 32 Correlation between increased Th1 cells and altered glucose metabolism in DIO was found especially for patients with T2D, whereas in patients with obesity this correlation is less evident.33 34 During DIO, the fraction of CD4+ effector T cells increases significantly, causing local inflammation by secretion of inflammatory cytokines in the adipose tissue. However, these pro-inflammatory effector T cells also migrate to other peripheral sites, such as the liver, further inducing inflammation.35 36 The increase in effector T cells is mediated by lymphocyte central memory cells. Central memory cells respond to inflammation and obesity-induced metabolic stress by proliferating and attaining an effector memory profile,37 contributing in part to the increase in T cells in adipose tissue during DIO.7 38 39 In our CD4CreCD40Lfl/fl mice, we observe a decrease in effector T cells in spleen and lymphoid organs, which is accompanied by a decrease in the expression of the pro-migratory chemokine receptor CXCR3. This suggests that absence of T cell CD40L affects migration of effector T cells into the periphery.40 However, in the adipose tissue the amount of effector T cells was unaffected when CD40L was absent on T cells. T lymphocyte central memory cells were also unaffected in the adipose tissue of CD4CreCD40Lfl/fl mice and these cells could thereby maintain the local effector memory populations. The decrease in CXCR3 expression does hamper effector T cells migratory ability, decreasing circulating effector cells and protecting peripheral organs such as the liver from these pro-inflammatory cells. As such, our data suggest that T cell CD40L deficiency does not limit the local expansion of the effector T cell, a culprit behind increased adipose tissue inflammation.
To maintain adipose tissue homeostasis Tregs are essential.31 32 These adipose tissue Tregs are predominantly induced Tregs (iTregs), which develop from naïve T cells, whereas natural Tregs (nTregs) develop in the thymus and largely depend on IL2 production.41 42 The CD4CreCD40Lfl/fl mice had decreased Treg numbers in blood, LNs, and spleen, but Treg numbers were maintained in the adipose tissue. This is probably due to the difference in development between the nTreg and iTreg. Blocking CD40L on lymphocytes leads to decreased IL2 production, as well as impaired de novo production of nTregs.29 The CD4CreCD40Lfl/fl mice seem to have a decrease in nTreg development due to T cell CD40L deficiency and reduction in IL2 expression online supplementary figure S1D). Adipose tissue iTregs are largely dependent on the B7 co-stimulatory pathway and peroxisome proliferator-activated receptor-γ (PPARγ).41 42 The B7 co-stimulatory molecules (CD80/CD86) on APCs play a critical role in iTreg differentiation, expansion, and function in adipose tissue, partly via induction of PPARγ in the differentiating naïve T cell.41 42 It is conceivable that these adipose tissue iTregs balance the local inflammation in our CD4CreCD40Lfl/fl mice. However, the amount of iTreg cells significantly decreases in DIO, and cross-sectional studies in humans have shown Treg decline to coincide with a decrease in B7 expression during obesity.9 41 This indicates the dependence of iTregs on the B7 proteins, rather than on CD40L, which is increased in DIO. We assume that in the T cell CD40L deficient mice, nTreg development was negatively affected, while iTreg development in adipose tissue was unaffected by CD40L deficiency, but iTreg cell numbers still decreased due to DIO.
Although the fraction of T cells seems to decrease in the CD4CreCD40Lfl/fl mice, we found an increase in the B cell fraction. On the contrary, total plasma IgG immunoglobulins were significantly decreased (online supplementary figure S7). This resembles findings from CD40L knock-out mice, where it was found that pro-inflammatory anti-oxidized lipid IgGs were reduced, correlating to an ameliorated metabolic phenotype in DIO.23 However, as we find limited changes in the metabolic syndrome of our T cell CD40L deficient mice, the increase in B cells and decrease in plasma IgGs do not seem to indicate biological relevance in our DIO model.
Apart from the shifts observed in immune cells, deficiency of T cell CD40L caused a more favorable metabolic and inflammatory profile in livers of obese mice, with a reduction in genes involved in lipid synthesis, a slight decrease in hepatosteatosis, a decrease in plasma insulin levels, and an altered immune cell composition. Gene expression levels of IL2 and IL12 were reduced in the liver, indicating reduced T cell proliferation.35 This was corroborated by a decrease in CD3+ expression, indicative of the decrease in effector T cells and Tregs. On the other hand, gene expression of pro-inflammatory IL6 was increased in the liver. However, data on the hepatocyte-specific deletion of the IL6 receptor in mice have shown to induce early onset insulin resistance and hepatosteatosis in DIO,43 demonstrating the protective function of IL6 in liver metabolism.
It seems that T cell CD40L orchestrates inflammation and metabolism via multiple mechanisms, although it does appear that the net result may be tissue dependent, of which the consequence seems to indicate a limited influence of T cell CD40L in diet-induced metabolic dysfunction on a whole. This reiterates the complicated role of the CD40-CD40L inflammatory dyad in DIO, where it has presented itself as a protective as well as detrimental mediator of obesity-associated metabolic syndromes.13–16 19 The most plausible explanation for the opposing phenotypes caused by CD40 and CD40L deficiency during DIO, is the presence of CD40 and CD40L on an abundance of cell types, all playing different roles in obesity. This has already become evident in previous studies. Although we and others found that total body CD40 deficiency was detrimental in DIO,13–16 we recently found that macrophage-specific CD40 deletion (LysMCreCD40fl/fl mice) had no effect on obesity-induced inflammation or metabolic dysregulation.24
Moreover, besides cell type specific effects, CD40 can also use different signaling routes via its TNF receptor-associated factors (TRAF) adaptor molecules to exert its effects. Obese mice deficient in CD40-TRAF2/3/5 signaling had aggravated adipose tissue inflammation and increased hepatosteatosis, whereas CD40-TRAF6 deficient obese mice had a decrease in adipose tissue inflammation and hepatosteatosis.13 Similar disparate effects are seen for the T cell co-stimulatory molecules CD137-CD137L, which are also expressed on an abundance of different cell types. Both inhibition of CD137 as well as antibody stimulation of CD137 gave similar results in DIO.44 45 These, sometimes contradicting results demonstrate the disparate effects co-stimulatory molecules may have in DIO, while highlighting the importance of understanding the effects of the co-stimulatory molecules on different cell types.
To conclude, T cell-specific CD40L is essential for effector T cell and Treg development, which are both important mediators in regulating obesity-associated inflammation. However, due to the opposing nature of these cells, T cell CD40L has limited effects on diet-induced metabolic dysregulation. Overall, T cell-specific CD40L deficiency only resulted in a slight improvement in insulin resistance, hepatic lipid uptake and hepatic inflammation, and therefore does not effectively reduce metabolic dysfunction and inflammation in obesity. As blocking CD40L in general constitutes a potent therapeutic strategy to reduce obesity-induced inflammation and its associated metabolic dysfunction,14 further research into the function of other CD40L expressing cell types, including the endothelial cell and the platelet, in DIO is warranted.
References
Footnotes
SABMA and EL contributed equally.
Contributors The study presented here was carried out in collaboration among all authors. MER contributed to concept and design, experiments and procedures, analysis and interpretation of data, statistical analysis and drafting of the manuscript. MdT, LW, BvO and MJJG performed experiments and acquired data. NG contributed to concept and design, and critical revision of the manuscript. SABMA contributed to concept and design, experiments and procedures, interpretation of data and critical revision of the manuscript. EL contributed to concept and design, data interpretation, critical revision of the manuscript for important intellectual content and obtained funding.
Funding This work was financially supported by The Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands, Organization for Health Research and Development and the Royal Netherlands Academy of Sciences for the GENIUS-II project ‘Generating the best evidence-based pharmaceutical targets for atherosclerosis-II’ (CVON). This study was also supported by the Netherlands Organization for Scientific Research (NWO) (VICI grant 016.130.676 to EL), the EU (H2020-PHC-2015-667673, REPROGRAM to EL), the European Research Council (ERC consolidator grant CD40-INN 681492 to EL), the German Science Foundation (DFG, CRC1123, project A5).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All experimental procedures were approved by the Ethical Committee for Animal Experiments of the Amsterdam UMC animal permit number (AVD1180020171666).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.