Article Text

Abstract
Objective Pancreatic beta cells proliferate in response to metabolic requirements during pregnancy, while failure of this response may cause gestational diabetes. A member of the vascular endothelial growth factor family, placental growth factor (PlGF), typically plays a role in metabolic disorder and pathological circumstance. The expression and function of PlGF in the endocrine pancreas have not been reported and are addressed in the current study.
Research design and methods PlGF levels in beta cells were determined by immunostaining or ELISA in purified beta cells in non-pregnant and pregnant adult mice. An adeno-associated virus (AAV) serotype 8 carrying a shRNA for PlGF under the control of a rat insulin promoter (AAV–rat insulin promoter (RIP)–short hairpin small interfering RNA for PlGF (shPlGF)) was prepared and infused into mouse pancreas through the pancreatic duct to specifically knock down PlGF in beta cells, and its effects on beta-cell growth were determined by beta-cell proliferation, beta-cell mass and insulin release. A macrophage-depleting reagent, clodronate, was coapplied into AAV-treated mice to study crosstalk between beta cells and macrophages.
Results PlGF is exclusively produced by beta cells in the adult mouse pancreas. Moreover, PlGF expression in beta cells was significantly increased during pregnancy. Intraductal infusion of AAV–RIP–shPlGF specifically knocked down PlGF in beta cells, resulting in compromised beta-cell proliferation, reduced growth in beta-cell mass and impaired glucose tolerance during pregnancy. Mechanistically, PlGF depletion in beta cells reduced islet infiltration of trophic macrophages, which appeared to be essential for gestational beta-cell growth.
Conclusions Our study suggests that increased expression of PlGF in beta cells may trigger gestational beta-cell growth through recruited macrophages.
- gestational diabetes mellitus
- placenta
- macrophage
- beta-cell growth
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
The expression and function of placental growth factor (PlGF) in the endocrine pancreas have not been studied before.
What are the new findings?
PlGF is exclusively expressed by beta cells in adult pancreas.
Increased beta cell-derived PlGF promotes gestational beta-cell growth through macrophages.
How might these results change the focus of research or clinical practice?
The contribution of insufficient beta cell-derived PlGF to the development of gestational diabetes deserves further investigation in the clinic.
Introduction
During pregnancy, the increase in maternal blood glucose requires the pancreas to produce more insulin to keep normal glucose homeostasis,1 and failure of this compensation may cause gestational diabetes.2 Although the high blood sugar in gestational diabetes usually returns to normal after pregnancy, previous studies have shown that gestational diabetes is a predisposing factor for type 2 diabetes, which occurs later in the life of pregnant women.3 Hence, understanding the etiology and pathogenesis of gestational diabetes is extremely important, given that this disease affects up to 10% of pregnant women.4
Studies on pregnant women and rodents have suggested that the gestational increases in insulin production and secretion by beta cells may be largely attributable to beta-cell growth during this period.5–10 Indeed, numerous studies have shown that gestational increase in beta-cell mass primarily stems from beta-cell proliferation.11–14 Failure to adequately increase beta-cell mass may cause glucose intolerance or even gestational diabetes. To date, the molecular mechanisms underlying gestational beta-cell growth are not fully understood.5–10
The intimate relationship between beta cells and endothelial cells starts during embryonic pancreatic organogenesis15 and plays a critical role in both endocrine pancreas development and beta-cell function in adults.16 This interaction is mainly mediated by vascular endothelial growth factor A (VEGF-A), the best studied ligand from the vascular endothelial growth factor (VEGF) family.17 We have previously shown that VEGF-A plays a role in the control of beta-cell mass in response to glucose alterations in the circulation,18 while the two major sources of VEGF-A in pancreas are beta cells and duct cells, which differentially regulate VEGF-A release.19 These earlier studies contributed to our understanding of the role of VEGF-A in beta-cell biology. However, VEGF family members other than VEGF-A biology have been minimally studied in beta-cell biology.
Placental growth factor (PlGF) is another member of the VEGF family and is thought to play a role in altered metabolic states or under some pathological circumstance.20 The expression and function of PlGF in the endocrine pancreas have not been examined. Here, we investigated PlGF expression in beta cells, as well as its role in gestational beta-cell growth.
Materials and methods
Production of AAV8 expressing shPlGF under a rat insulin promoter (RIP)
A PlGF shRNA sequence (GCTGTTCACTTGCTTCTTAGTCGAGTAAGAAGCAAGTGAACAGC) or a scramble (GCTGAGTACTTCGAAATGTCGTCGAGGACATTTCGAAGTACTCAGCG) was designed using Block-IT RNAi designer and was cloned into pAAV-mcherry-flex-dtA (dTA) (Addgene, No 58536, Cambridge, Massachusetts, USA) by restriction sites BsrG1 and SalI. A RIP described before21 was swapped into the vector by restriction sites FseI and XbaI. Adeno-associated virus (AAV) serotype 8 vectors were generated by cotransfection with the shPlGF or the scrambled plasmid, a packaging plasmid carrying rep and cap from the AAV serotype 8, and a helper plasmid carrying the adenovirus helper in human embryonic kidney 293 cells, as described before.22 AAV viruses were purified by polyethylene glycol/aqueous two-phase partitioning and then stored at −80°C for later use. Titration of viral vectors was determined by an AAVpro Titration kit (TaKaRa Bio, Mountain View, California, USA). Recombinant mouse PlGF was purchased from Abcam (Ab207150, Cambridge, Massachusetts, USA) and was delivered to mice via pancreatic duct at 20 µg/mouse. The control mice received saline. Transfection of a beta-cell line Min6 (American Type Culture Collection, Rockville, Maryland, USA) was performed using Lipofectamine 3000 (Invitrogen, Carlsbad, California, USA). Min6 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen) supplied with 10% fetal bovine serum (Invitrogen), 2 mmol/L glutamine at 37°C, 95% air/5% CO2.
Mouse manipulations
The BAC transgenic elastase promoter CreERT reporter (Ela-CreERT) mouse was described before.19 C57BL/6, Rosa26CAGTomato (Tomato)18 and INS1cre knock-in23 mice were all purchased from the Jackson Lab (Bar Harbor, Maine, USA). Tamoxifen induction of Tomato expression in acinar cells in Ela-CreERT; Tomato mice has been described before.19 INS1cre knock-in mice were bred with Tomato mice to generate INS1cre; Tomato mice for purification of beta-cells based on red fluorescence. Fasting blood glucose monitoring and intraperitoneal glucose tolerance testing were performed as described before.21 Pancreatic intraductal virus infusion was performed as described.24 Injection of 200 µL chlodronate and control liposome (clodronate liposomes, Netherlands) from the tail vein of the mice was performed twice per week, starting 1 day before time breeding until the end of the experiment, as previously described.21
RNA isolation and quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted using the RNeasy mini kit (Qiagen, Valencia, California, USA) and then quantified with the Nanodrop 1000 (Thermo Fisher Scientific, Waltham, Massachusetts, USA), followed by cDNA synthesis (Qiagen) and RT-qPCR. Primers for CycloA (QT00247709) and PlGF (QT00103222) were both purchased from Qiagen. Relative values of mRNA levels were obtained by sequential normalization against the housekeeping gene cyclophilin A and the experimental control.
PlGF ELISA
PlGF levels were determined with a PlGF ELISA kit (R&D Systems, Los Angeles, California, USA). All reagents, standard dilutions and samples were prepared as directed in the product insert. First, 100 µL of Assay Diluent was added to each well, followed by 100 µL of standard, control, or sample to each well. The microplate was then covered with a plate sealer and incubated at room temperature for 2 hours. Each well was aspirated and washed three times, after which 200 µL of conjugate was added to each well. The microplate was then covered with a new plate sealer and incubated at room temperature for 1 hour before another four times of aspiration and wash. Finally, 200 µL substrate solution was added to each well, and the microplate was incubated at room temperature for 30 min before 50 µL of stop solution was added to each well. The microplate was read at 450 nm within 30 min. The wavelength correction was set to 540 or 570 nm.
Pancreatic digestion and FACS
Pancreatic duct perfusion, pancreas digestion, islet isolation, single islet cell preparation, fluorescence-activated cell sorting (FACS) and confirmation of the sorted cell purity were performed as described before.19
Immunohistochemistry, quantification of proliferating beta cells and beta-cell mass
All pancreas samples were fixed and cryo-protected in 30% sucrose overnight before freezing, as described before.25 Fast red staining was performed with the chromogen system (Dako, Carpinteria, CA, USA). For fluorescent staining, mCherry was detected by direct fluorescence. Primary antibodies used were: guinea pig polyclonal anti-insulin (Dako), goat polyclonal anti-PlGF (R&D Systems), rat polyclonal anti-Ki-67 (Dako) and rat anti-F4/80 (Invitrogen). Secondary antibodies were all purchased from Dako. Nuclear staining (Hoechst 33342 (HO)) was performed with Hoechst 33 342 (Becton-Dickinson Biosciences, San Jose, California, USA). Quantification of beta-cell proliferation was based on the percentage of Ki-67+ cells in total beta cells, as previously described.26 Beta-cell mass was analyzed as described before.21 Briefly, the mouse pancreata were trimmed of all non-pancreatic tissue, weighed, fixed, cryoprotected in 30% sucrose overnight before freezing in a way to allow longitudinal sections from tail to head of the pancreas to be obtained. Sections at 150 µm intervals from whole pancreas were immunostained for insulin. The beta-cell mass per pancreas was estimated as the product of the relative cross-sectional area of beta cells per total tissue and the weight of the pancreas.
Data analysis
For in vivo experiments, five mice were used for each group. All data were statistically analyzed by one-way analysis of variance with a Bonferroni correction, followed by Fisher’s exact test. All error bars represent SD. Significance was presented as * when p<0.05. No significance was presented as NS. P value and n value are indicated in the figure legends.
Results
PlGF is exclusively produced by beta cells in the adult mouse pancreas
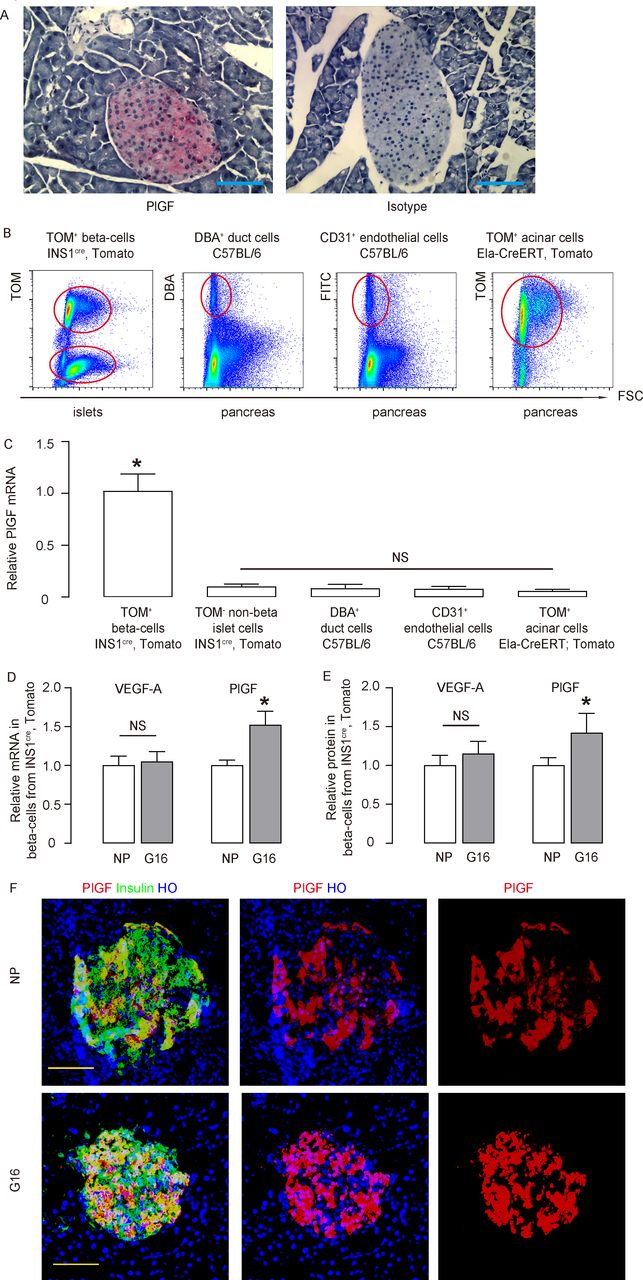
VEGF-A in beta cells plays an important role in the control of beta-cell development and function, while the role of PlGF in beta cells is unknown. Here, we found that in the mouse pancreas, PlGF+ cells were exclusively located in the islet (figure 1A). We compared PlGF transcripts among different FACS-purified pancreatic cell types (beta cells (red fluorescent cells from islets of INS1cre; Tomato mice), non-beta-cells (non-red fluorescent cells from islets of INS1cre; Tomato mice), duct cells (Dolichos biflorus agglutinin (DBA)+ cells from C57BL/6 mice), endothelial cells (CD31+ cells from C57BL/6 mice) and acinar cells (Tomato+ cells from tamoxifen-treated Ela-CreERT; Tomato mice)) (figure 1B). We found that beta cells were the only pancreatic cell type that expressed significant PlGF (figure 1C).

PlGF expresses in the adult beta cells and upregulates during pregnancy. (A) Immunostaining for PlGF (fast red) in normal mouse pancreas, compared with the isotype antibody control. (B) Representative FACS charts to show purification of beta cells (red fluorescent cells from islets of INS1cre, tomato mice), non-beta cells (non-red fluorescent cells from islets of INS1cre, tomato mice), duct cells (DBA+ cells from C57BL/6 mice), endothelial cells (CD31+ cells from C57BL/6 mice) and acinar cells (tomato+ cells from tamoxifen-administrated Ela-CreERT, tomato mice). (C) RT-qPCR for PlGF mRNA levels among different FACS-purified pancreatic cell types. (D,E) VEGF-A and PlGF mRNA and protein levels were determined in purified beta cells from INS1cre, tomato mice, by RT-qPCR (D) and by ELISA (E), respectively, in the NP mice and litter mate pregnant mice at G16. (F) Representative immunostaining for PlGF and insulin in the NP and G16 mouse pancreas. *p<0.05. n=5. Scale bars are 50 µm. DBA, dolichos biflorus agglutinin; Ela-CreERT, elastase promoter CreERT reporter; FACS, fluorescence-activated cell sorting; FITC, fluorescein isothiocyanate; G16, gestational day 16; HO, Hoechst 33342; mRNA, messenger RNA; NP, non-pregnant; NS, non-significant; PlGF, placental growth factor; RT-qPCR, quantitative reverse transcription PCR; TOM, tomato; VEGF-A, vascular endothelial growth factor A.
PlGF in beta cells upregulates during pregnancy
It is well known that pancreatic beta cells proliferate in response to metabolic needs during pregnancy, while failure of this response may lead to gestational diabetes. Since beta cells produce both VEGF-A and PlGF, we then examined expression of both VEGF-A and PlGF in beta cells in pregnant INS1cre; Tomato mice. Interestingly, we found that VEGF-A levels in beta cells remain unchanged during pregnancy but significantly increased at gestational day 16 (G16, the peak of beta-cell proliferation in pregnant mice),14 compared with non-pregnant (NP) litter mates, by RT-qPCR (figure 1D) and by ELISA (figure 1E). PlGF and insulin coimmunostaining was done on pancreas from NP and G16 mice, and both showed beta cell-specific PlGF expression (figure 1F). The PlGF signals appeared to be higher in G16, compared with NP (figure 1F). These data suggest that PlGF may play a role in gestational beta-cell growth. VEGFR1 is the unique receptor for PlGF, and VEGFR1 is expressed in both macrophages and endothelial cells, but not in any endocrine cells.19 27–30 Thus, we examined the levels of VEGFR1 in sorted islet macrophages and endothelial cells. We did not detect alteration in VEGFR1 levels on these cells (online supplementary figure 1), suggesting that VEGFR1 expression level is not a cause for increased beta cell-derived PlGF during pregnancy.
Supplemental material
Beta cell-specific knockdown of PlGF
In order to evaluate the role of PlGF during gestational beta-cell growth, we used AAV serotype 8 carrying a short hairpin small interfering RNA for PlGF (shPlGF) under the control of a RIP to specifically deplete PlGF in beta cells (AAV–RIP–shPlGF). An AAV carried a scrambled sequence under RIP (AAV–RIP–scrambled) as controls. An mCherry reporter was also included in the AAV backbone to allow the transduced cells to be visualized by red fluorescence (figure 2A). The mouse islet endothelial cell line MS1 (a non-beta-cell line used as a control) and mouse insulinoma cell line Min6 were both transduced by AAV–RIP–shPlGF (figure 2B). PlGF mRNA was significantly knocked down in Min6 cells but was unaltered in MS1 cells, confirming the specificity of RIP and efficient knockdown of shPlGF in AAV–RIP–shPlGF (figure 2C). Moreover, PlGF protein in the cellular extracts and in conditioned media from AAV–RIP–shPlGF-transduced Min6 cells was reduced by more than 70% (figure 2D).

Beta cell-specific knockdown of PlGF. (A) In order to evaluate the role of PlGF during gestational beta-cell growth, we used AAV serotype 8 carrying an shPlGF under the control of an RIP to specifically inhibit PlGF production in beta cells (AAV–RIP–shPlGF). A control AAV carried a scrambled sequence under RIP (AAV–RIP–scrambled). An mCherry reporter was also included in the AAV backbone to allow transduced cells to be visualized by red fluorescence. (B,C) RT-qPCR for PlGF in MS1 (B) and MIN6 (C) cells transduced with AAV–RIP–shPlGF or AAV–RIP–scrambled. (D) ELISA for PlGF in cellular compartment or conditioned media in AAV–RIP–shPlGF–transduced MIN6 cells. *p<0.05. n=5. AAV, adeno-associated virus; NS, non-significant; PlGF, placental growth factor; RIP, rat insulin promoter; RT-qPCR, quantitative reverse transcription PCR; shPlGF, short hairpin small interfering RNA for PlGF.
Depletion of PlGF in beta cells impairs the glucose response during pregnancy
The AAV–RIP–shPlGF or AAV–RIP–scrambled virus was infused into mouse pancreas from the pancreatic duct to specifically knock down PlGF in beta cells in 10-week-old female C57BL/6 mice as described.21 25 31 Three weeks later, the incision on these mice was healed, and some mice were analyzed, showing no difference in beta-cell proliferation and beta-cell mass between two experimental groups (online supplementary figure 2). Other mice were time-mated with male C57BL/6 mice. The pregnant mice were sacrificed at G16, the peak of beta-cell proliferation as reported by us14 and by others5–10 (figure 3A). At G16, we detected red fluorescent cells only in beta cells (about 80% of beta cells were red) from the mice that had received either AAV–RIP–shPlGF or AAV–RIP–scrambled, suggesting that these viruses only transduced beta cells, which again confirmed the specificity of the RIP in the viral structure (figure 3B–C). Moreover, PlGF expression was greatly attenuated in beta cells from AAV–RIP–shPlGF-treated mice, compared with beta cells from AAV–IP–scrambled-treated mice (figure 3D). Quantification of PlGF protein in isolated mouse islets showed more than 50% reduction in PlGF (figure 3E), suggesting successful knockdown of PlGF in beta cells in AAV–RIP–shPlGF-treated mice. At G16, although the fasting glycemia remained unaltered in AAV–RIP–shPlGF-treated mice (figure 3F), these mice exhibited compromised glucose response (figure 3G) and reduced serum insulin (online supplementary figure 3A), compared with AAV–RIP–scrambled-treated mice. Moreover, insulin tolerance test (ITT) did not show difference among the two groups of mice (online supplementary figure 3B). Therefore, beta cell-specific knockdown of PlGF leads to impairment of glucose response during pregnancy, likely through compromised beta-cell growth rather than through reduced insulin release.

Depletion of PlGF in beta cells impairs the glucose response during pregnancy. (A) Schematic of in vivo study. The AAV–RIP–shPlGF or AAV–RIP–scrambled virus was infused into pancreatic duct to specifically knockdown PlGF in beta cells in female C57BL/6 mice. Three weeks later, these mice were time-mated with male C57BL/6 mice and sacrificed at G16. (B,C) Representative images for mCherry, insulin and HO staining (B), and quantification for % mCherry+ beta cells (C). (D) Quantification for islet PlGF protein levels by ELISA. (E) Representative images for PlGF, insulin and HO staining in G16 pregnant mouse pancreas. (F) Fasting glycemia at G16. (G) IPGTT for pregnant mice at G16. *p<0.05. Five mice were included in each experimental group. Scale bars are 50 µm. AAV, adeno-associated virus; G16, gestational day 16; HO, Hoechst 33342; IPGTT, intraperitoneal glucose tolerance testing; NS, non-significant; PlGF, placental growth factor; RIP, rat insulin promoter; shPlGF, short hairpin small interfering RNA for PlGF.
Beta cell-specific knockdown of PlGF inhibits beta-cell proliferation, resulting in attenuated gestational beta-cell growth
Next, we explored the mechanisms underlying the impairment of the glucose response in AAV–RIP–shPlGF-treated pregnant mice. Beta-cell proliferation was determined by Ki-67 positivity in beta cells, and the outcome of the alteration in beta-cell proliferation was assessed by beta-cell mass at G16. We detected a significant reduction in beta-cell proliferation in AAV–RIP–shPlGF-treated mice, compared with AAV–RIP–scrambled-treated mice, as shown by representative images (figure 4A) and by quantification (figure 4B). Moreover, the increases in beta-cell mass were attenuated in AAV–RIP–shPlGF-treated mice, compared with AAV–RIP–scrambled-treated mice (figure 4C). Thus, beta cell-specific knockdown of PlGF inhibits beta-cell proliferation, resulting in attenuated gestational beta-cell growth.
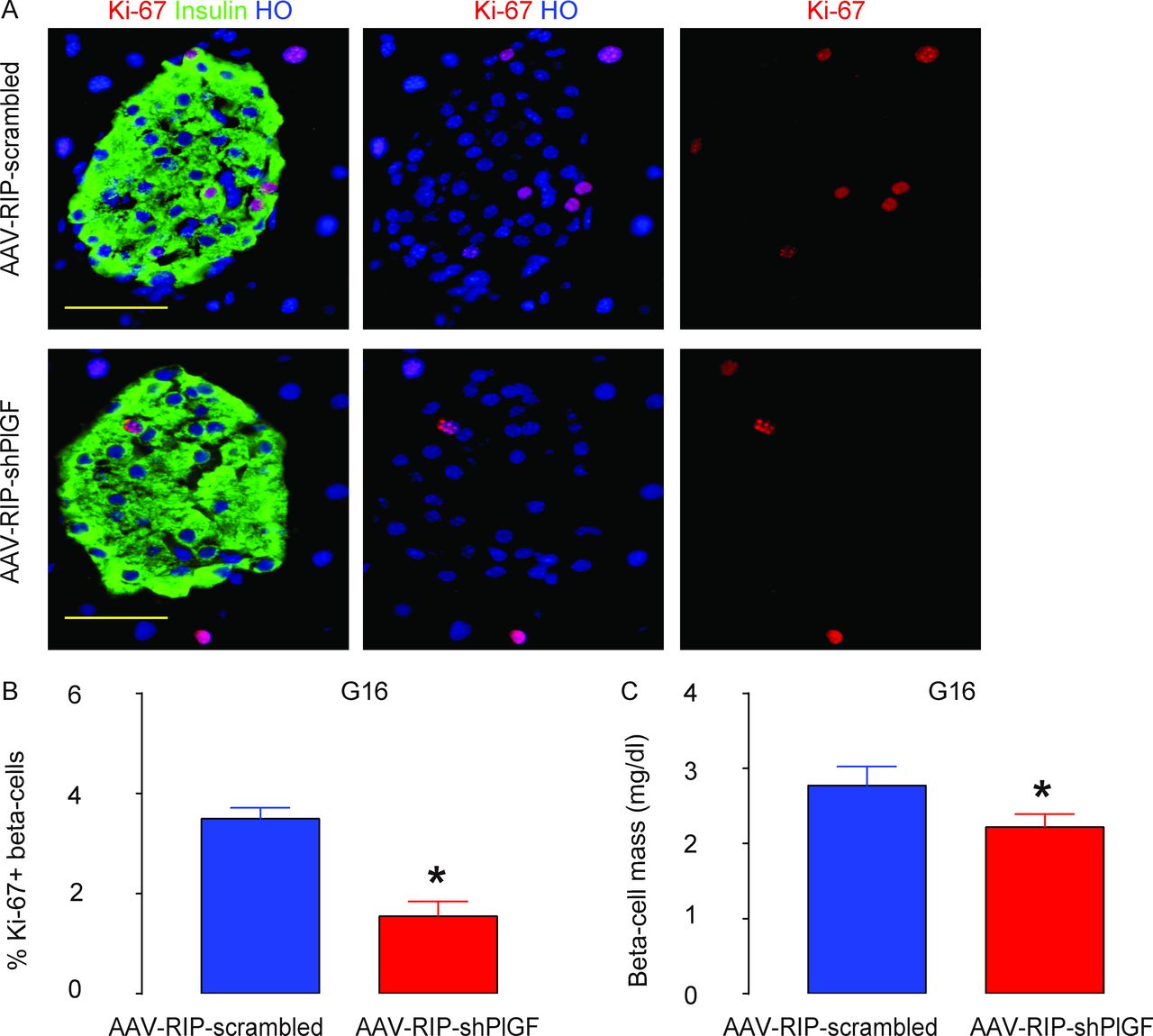
Beta cell-specific knockdown of PlGF inhibits gestational beta-cell proliferation, resulting in attenuated beta-cell growth during pregnancy. (A,B) Ki-67+ beta cells were analyzed in the pancreas from G16 pregnant C57BL/6 mice that had received either AAV–RIP–shPlGF or AAV–RIP–scrambled, shown by representative images (A) and by quantification (B). (C) Beta-cell mass. *p<0.05. Five mice were included in each experimental group. Scale bars are 50 µm. AAV, adeno-associated virus; G16, gestational day 16; HO, Hoechst 33342; RIP, rat insulin promoter; shPlGF, short hairpin small interfering RNA for PlGF.
PlGF promotes gestational beta-cell proliferation through recruitment of macrophages
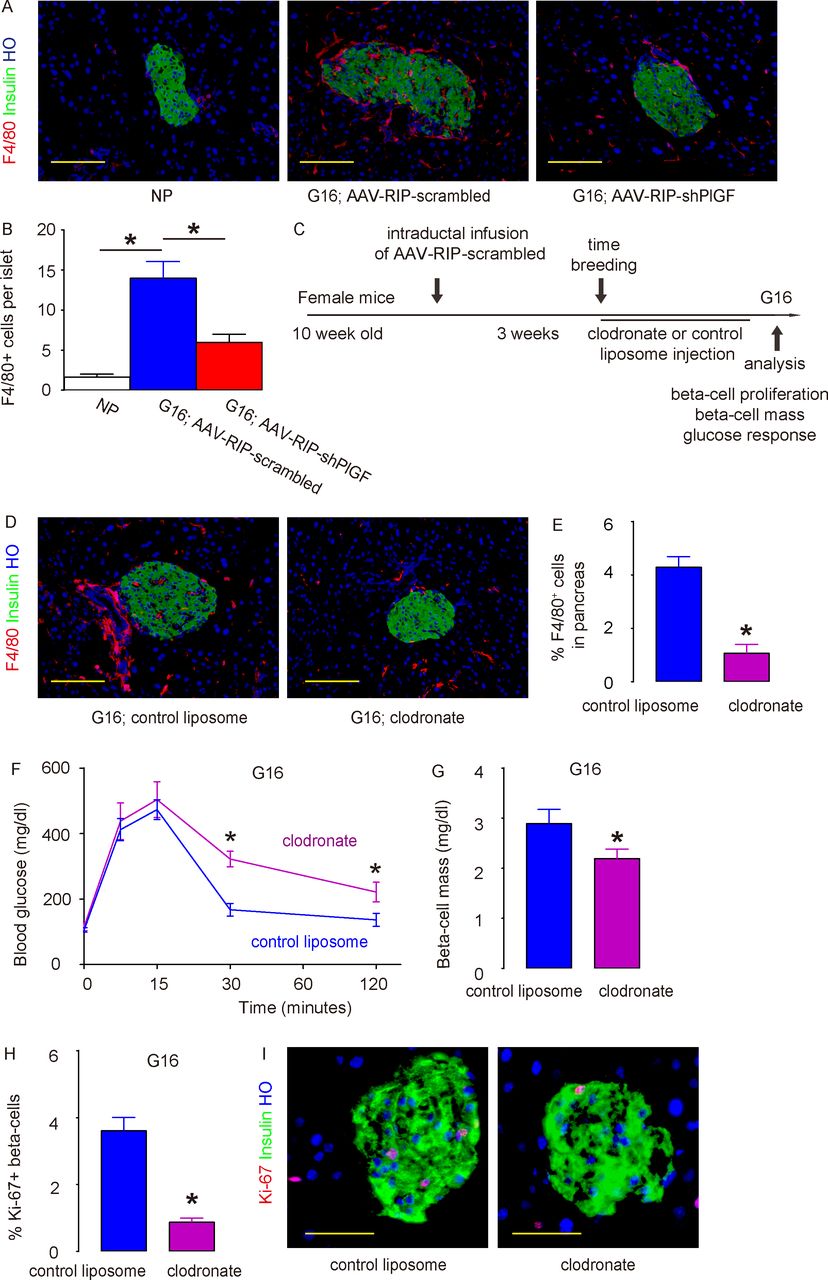
Recently, we and others have shown a role of macrophages in beta-cell proliferation and regeneration.16 21 32–34 As mentioned earlier, PlGF was a potent ligand for VEGFR1, which is expressed by macrophages.35–37 Thus, we hypothesized that the effects of beta cell-derived PlGF on beta-cell proliferation may be mediated by macrophages. To prove it, we examined the recruitment of macrophages to the mouse islets in virus-treated pregnant mice at G16. F4/80 is a pan-macrophage marker. While very few F4/80+ macrophages were detected in the NP mouse pancreas, many more macrophages were found inside and close to the islets in pregnant mice that had received AAV–RIP–scrambled, while this increase in macrophage infiltration into the islet region in pregnant mice was greatly reduced in AAV–RIP–shPlGF-treated mice (figure 5A,B). To prove that this increase in macrophages by PlGF in beta cells contributes to gestational beta-cell growth, we used a macrophage-depleting reagent, clodronate, in AAV–RIP–scrambled-treated pregnant mice. The control mice received control liposome instead of clodronate (figure 5C). At G16, we detected a significant reduction in pancreatic macrophages (including islet region) in mice that had received clodronate, compared with those that had received control liposome (figure 5D,E). Clodronate treatment did not cause overt diabetes but induced poorer glucose response (figure 5F), likely resulting from impaired beta-cell growth (figure 5G). The impaired beta-cell growth likely resulted from reduced beta-cell proliferation (figure 5H,I). In order to further clarify the role of PlGF in gestational beta-cell proliferation, we injected recombinant mouse PlGF or control saline solution from the pancreatic duct and examined beta-cell proliferation and intraislet macrophages after 7 days. We detected significant increases in Ki-67+ beta cells and significant increases in islet F4/80+ macrophages (online supplementary figure 4). Furthermore, PlGF did not alter beta-cell proliferation in vitro (1.5%±0.2% Ki-67+ beta cells by PlGF vs 1.4%±0.2% by control saline), reinforcing our conclusion for an importance role of macrophages in PlGF-mediated gestational beta-cell growth.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PlGF promotes gestational beta-cell proliferation through recruitment of macrophages. (A,B) Representative images for immunostaining for F4/80 and insulin in NP mouse pancreas, pregnant mouse at G16 treated with AAV–RIP–shPlGF or AAV–RIP–scrambled virus (A) and quantification for intraislet macrophage number (B). (C) Schematic of macrophage depletion study. The AAV–RIP–scrambled virus was infused into the pancreatic duct in female C57BL/6 mice. Three weeks later, these mice were time-mated with male C57BL/6 mice and received either clodronate or control liposome until sacrifice at G16. (D,E) Representative images for F4/80, insulin and HO staining (D) and quantification of F4/80+ cells in pancreas (E). (F) IPGTT for pregnant mice at G16. (G) Beta-cell mass. (H,I) Ki-67+ beta cells were analyzed in the pancreas from G16 pregnant C57BL/6 mice that had received either clodronate or control liposome, shown by quantification (H) and by representative images (I). *p<0.05. Five mice were included in each experimental group. Scale bars are 50 µm. AAV, adeno-associated virus; G16, gestational day 16; HO, Hoechst 33342; IPGTT, intraperitoneal glucose tolerance testing; NP, non-pregnant; RIP, rat insulin promoter; shPlGF, short hairpin small interfering RNA for PlGF.
Discussion
The insulin need increases during pregnancy and is met by increases in insulin production, release and sensitivity.1 Failure of this compensation may lead to glucose intolerance or even gestational diabetes, which has been shown to predispose the development of type 2 diabetes.3 Adequate gestational beta-cell growth is critical for maintaining gestational glucose metabolism, which relies on proper production and secretion of pregnancy-associated growth factors.38
Beta cells release many factors directly to pancreas in a paracrine manner, and these factors may act as chemoattractants to recruit the cells that express the corresponding receptors.39 In the current study, PlGF is such a chemoattractant ligand that is produced and secreted exclusively by beta cells in pancreas, while pancreatic macrophages express VEGFR1, the unique receptor for PlGF.20 Although the levels of PlGF in beta cells increased less than twofolds, it is known that the physiological level of a VEGF family member is maintained in a relatively small range,17 40 and a modest change may exert significant biological changes.41 42 VEGF-A may also play a role in the PlGF-associated effects since VEGF-A regulates vessel permeability, binds VEGFR1 similarly like PlGF, and even forms heterodimers with PlGF.43 44 A delicate interaction between VEGF-A and PlGF may coordinate the recruitment of pancreatic macrophages into the islet niche in a tightly organized manner, likely involving multidirectional crosstalk among beta cells, islet endothelial cells and pancreatic macrophages.27 Of note, we have previously shown that VEGFR1 is also expressed in islet endothelial cells.18 Hence, PlGF/VEGFR1 signaling may also regulate the restructure of islet endothelia necessary for circulated monocytes/macrophages to enter the islet niche and to join to the tissue-resident macrophages.
The major source of PlGF during pregnancy is the placenta.45 It is well known that beta cells locate in a niche where they directly contact with islet endothelial cells.15 The placenta-derived PlGF in circulation needs to cross the vascular endothelial cells to enter pancreatic parenchyma and attract VEGFR1-expressing macrophages in pancreas. Due to the expression of VEGFR1 on vascular endothelial cells, the majority of placenta-derived PlGF will be ‘consumed’ in the endothelial cells before entering the pancreatic zone. On the other hand, the beta cell-derived PlGF is likely released into pancreatic parenchyma in a paracrine manner, which attracts pancreatic macrophages that harbor the VEGFR1.
The basal expression of PlGF is much lower than VEGF-A in beta cells. PlGF does not likely play a critical role in baseline postnatal beta-cell homeostasis, since infusion of AAV–RIP–shPlGF did not alter beta-cell mass and glucose response of the mice. However, PlGF did play a critical role in postnatal beta-cell growth in response to increased metabolic need during pregnancy. The increase in PlGF levels in beta cells during pregnancy is only about 50%. However, previous studies have shown that the biological effects of the angiogenic factors are extremely narrow.17 40–42 For example, inactivation of only one allele of VEGF-A resulted in embryonic lethality at midgestation,46 47 while a twofold increase in VEGF-A levels also led to embryonic lethality.48 Therefore, the seemingly modest increase in PlGF levels in beta cells can exert a significant biological effect. The reduction in gestational growth in beta-cell mass by shPlGF was significant but not huge. Nevertheless, it did result in impaired glucose response, suggesting that the typical growth in beta-cell mass during pregnancy may be immediately enough for maintaining proper metabolic requirement without much reserve, which is consistent with the clinical reports showing that some women with gestational diabetes became normoglycemic after pregnancy.2
The intraductal infusion technique allows the pancreas-specific delivery of AAVs, as we have demonstrated in previous reports.19 21 24 25 31 49 The AAV-mediated beta cell-specific depletion of PlGF has an advantage over use of beta cell-specific PlGF knockout mice, which are even not available worldwide. Using RIP-Cre to knock out PlGF in beta cells will developmentally alter beta-cell phenotype. On the other hand, using RIP-creERT to knock out PlGF cannot bypass using tamoxifen, which binds to and signals through estrogen receptor highly expressed in beta cells to cause off-target effects.50 51 Although here we have shown that PlGF is exclusively produced by beta cells among all pancreatic cells, some inflammatory cells in the pancreas also express certain levels of PlGF.52 Thus, our use of the insulin promoter to drive expression of shPlGF assured the absence of effects from the knockdown of PlGF in non-beta cells.
The beta-cell-trophic factors that are produced and secreted by macrophages will be interesting targets to be studied in the future. Previous studies have suggested that WNT ligands,32 33 VEGF-A,16 and combined epidermal growth factor and transforming growth factor β121 may be such candidates. These studies also highlighted M2 macrophages, an alternatively polarized subtype of macrophages (as opposed to the classical M1 macrophages), as a very important regulator of beta-cell proliferation. It would thus not be surprising if the recruited macrophages to the beta cells during pregnancy are predominantly M2 macrophages.
MIP-GFP (mouse insulin promoter driving a green fluorescent protein reporter) mice have been commonly used for purification of beta cells.53 However, recent studies showed that the presence of human growth hormone (hGH) in the transgenic mice, including MIP-GFP, appeared to affect beta-cell function and proliferation through prolactin receptor signaling.54 55 Thus, INS1cre; Tomato mice, with the absence of hGH in either parental strain, were used to purify beta cells and non-beta islet cells in the current study.
Here, we presented a novel model for studying cell–cell communication in beta-cell proliferation. Our study suggests that increased expression of PlGF in beta cells may trigger gestational beta-cell growth through recruited macrophages. It may be intriguing to examine a possible contribution of insufficient beta cell-derived PlGF to the development of gestational diabetes in future studies.
References
Footnotes
Contributors WY, YJ, YW, TZ, QL, CW, GS, CC, NW, KP and GKG: acquisition and analysis of data; XX: study conception and design, funding, acquisition, analysis and interpretation of data, manuscript writing and guarantee of the study.
Funding This work was supported by tenure-track Assistant Professor Startup from Division of Pediatric Surgery of Children’s Hospital of Pittsburgh (to XX) and funding of Children’s Hospital of Pittsburgh of the UPMC health system (RAC, to XX), National Nature Science Foundation of China (grant number 81600599, to WY), Foundation for Young Scholars of Affiliated Hospital of Nantong University (grant number TDFY0303, to WY) and the project funded by the Priority Academic Program Development of Nantong Talent Center (grant number 2016-III-458, to WY).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All animal experiments were approved by the Institutional Animal Care and Use Committee at University of Pittsburgh.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. This does not include identifiable patient data.