Article Text

Abstract
Objective The present study focused on the development of a poloxamer 407 thermosensitive hydrogel loaded with keratinocyte growth factor-2 (KGF-2) and fibroblast growth factor-21 (FGF-21) as a therapeutic biomaterial in a scald-wound model of type-2 diabetes in Goto-Kakizaki (GK) rats.
Research design and methods In this study, a poloxamer 407 thermosensitive hydrogel loaded with KGF-2 and/or FGF-21 was prepared and its physical and biological properties were characterized. The repairing effects of this hydrogel were investigated in a scald-wound model of type-2 diabetes in GK rats. The wound healing rate, epithelialization, and formation of granulation tissue were examined, and biomarkers reflecting regulation of proliferation and inflammation were quantified by immunostaining and Western blotting. T tests and analyses of variance were used for statistical analysis via Graphpad Prism V.6.0.
Results A 17.0% (w/w) poloxamer 407 combined with 1.0% (w/w) glycerol exhibited controlled release characteristics and a three-dimensional structure. A KGF-2/FGF-21 poloxamer hydrogel promoted cellular migration without apoptosis. This KGF-2/FGF-21 poloxamer hydrogel also accelerated wound healing of scalded skin in GK rats better than that of a KGF-2 or FGF-21 hydrogel alone due to accelerated epithelialization, formation of granulation tissue, collagen synthesis, and angiogenesis via inhibition of inflammatory responses and increased expression of alpha-smooth muscle actin (α-SMA), collagen III, pan-keratin, transforming growth factor-β (TGF-β), vascular endothelial growth factor (VEGF), and CD31.
Conclusions A KGF-2/FGF-21 poloxamer hydrogel accelerated wound healing of scalded skin in GK rats, which was attributed to a synergistic effect of KGF-2-mediated cellular proliferation and FGF-21-mediated inhibition of inflammatory responses. Taken together, our findings provide a novel and potentially important insight into improving wound healing in patients with diabetic ulcers.
- type 2 diabetes
- wound healing
- combination therapy
- growth factor(s)
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Keratinocyte growth factor-2 (KGF-2) promotes epithelial repair and has been developed for ophthalmic preparations.
Fibroblast growth factor-21 (FGF-21) plays an important role in metabolic regulation, which has been shown to have a significant effect on lowering blood glucose levels.
What are the new findings?
A KGF-2/FGF-21 hydrogel accelerated the healing of scalded skin in Goto-Kakizaki rats better than that of a KGF-2 or FGF-21 hydrogel alone.
The superior KGF-2/FGF-21 hydrogel healing rate was attributed to a synergistic effect of KGF-2-mediated cellular proliferation and FGF-21-mediated inhibition of inflammatory responses.
How might these results change the focus of research or clinical practice?
Our findings suggest that the use of a KGF-2/FGF-21 hydrogel may represent an effective clinical treatment for promoting wound healing in patients with diabetic ulcers.
Introduction
Diabetic foot ulcers (DFU) are a chronic complication of diabetes and are the main cause of diabetes-related amputations, which not only induce considerable mental stress to patients but also seriously impair their quality of life and result in financial burdens.1 2 Although negative-pressure drainage, anti-infective dressing, and growth factors have been demonstrated to partially improve healing of DFUs,3 single-treatment methodologies focusing on either minimizing wound inflammation or promoting cellular proliferation have been unable to yield satisfactory results. Therefore, treatment methods and/or therapeutic compounds targeting multiple stages of the healing process of DFUs are hypothesized to be more beneficial for improving the healing rate of DFUs.
Wound repair is a complex yet orderly biological process that includes phases involving hemostasis, inflammatory reactions, cellular proliferation, and tissue remodeling.4–7 An issue during any one of these phases can delay wound healing. The hyperglycemic microenvironment in diabetic patients is likely to cause a decrease in the secretion of growth factors associated with healing, leading to a decrease in fibroblast proliferation,8 which is one of the most detrimental factors for delaying healing of diabetic ulcers. Members of the fibroblast growth factor (FGF) family play important roles in regulating wound repair, embryonic development, angiogenesis, and nerve repair.9 One such FGF member, keratinocyte growth factor-2 (KGF-2; also known as FGF-10), has been demonstrated to be critical in the wound healing process.10–12 Previous studies have demonstrated that when epithelial tissue is damaged, KGF-2 mRNA is overexpressed at the wound surface, stimulating the accumulation of fibroblasts, collagen, and connective tissue to the wound site to produce new tissue and promote wound healing.13 The ongoing Phase-III clinical trial of KGF-2 from our lab has revealed that KGF-2 significantly promotes the healing process in patients with second-degree burns. These aforementioned animal and human clinical-trial data led us to hypothesize that KGF-2 may be useful in the treatment of diabetes and other disease-induced chronic ulcers. However, in diabetic chronic ulcers, high glucose levels often induce the production of excessive inflammatory factors, leading to prolonged inflammatory responses, destruction of the wound microenvironment due to accumulation of inflammatory exudates, and cellular damage, all of which represent challenges to healing diabetic ulcers.14 15 A newly discovered member of the FGF family, FGF-21, has been shown to have a significant effect on lowering blood glucose levels.16–18 Moreover, recent studies have shown that FGF-21 exhibits an excellent efficacy in regulating inflammation.19–22 In terms of a corresponding mechanism of action, FGF-21 inhibits macrophage-mediated inflammatory responses by activating nuclear factor erythroid 2-related factor 2 and inhibiting the NF-kappa-B signaling pathway.23 Therefore, the use of FGF-21 to improve the inflammatory microenvironment, combined with KGF-2 to promote cellular proliferation, may represent an effective treatment for promoting wound healing in patients with diabetic ulcers.
One caveat is that, therapeutic growth factors, when applied on the wound surface, are often easily degraded by proteases secreted by the inflammatory microenvironment. This caveat represents the bottleneck for using growth-factor-like protein therapeutics for the treatment of wound repair. The rapid development of tissue engineering technologies in recent years has provided a basis for maximizing the pharmacological effects of these therapeutic agents. Tissue engineering biomaterials that are both biocompatible (no tissue rejection) and biodegradable function to induce tissue regeneration, promote cellular growth/proliferation, and are capable of slowly releasing growth factors on the wound surface, which prevents hydrolysis by proteases.24–26 Poloxamer 407 is a class of non-ionic surfactants under the trade name, Pluronic F127, and consists of monomeric units of polyoxyethylene and polyoxypropylene.27 Poloxamer 407 is considered to be one of the safest polymer materials for preparing thermosensitive hydrogel tissue-functional materials, and has been demonstrated to exhibit good biocompatibility and low irritation.26 The advantages of using poloxamer hydrogels for the treatment of chronic skin ulcers (such as DFUs) include their two-way permeabilities to water and gas,28 as well as them being effective barriers against bacterial invasion and infection.29 Additionally, poloxamer hydrogels exhibit good plasticity and adhesion,30–32 which prolong the residence time of the drug on the skin surface. Finally, poloxamer hydrogels form three-dimensional scaffolds that not only provide the matrix components needed for new wound healing26 but also function as local sites that slowly release growth factors.
Therefore, in the present study, we prepared thermosensitive hydrogels encapsulating KGF-2 and/or FGF-21 using poloxamer 407 and investigated their therapeutic effects in wound healing of scalded skin in diabetic rats, with an emphasis on both the safeties and efficacies of such treatments.
Methods
Preparation and characterization of poloxamer 407 hydrogels loaded with KGF-2 and FGF-21
According to our supplementary data, we chose a 17.0% (w/w) poloxamer 407 (Pluronic F127, BASF, Rheinland-Pfalz, Germany) combined with 1.0% (w/w) glycerol (Guangdong Guanghua Sci-Tech Co., China) as the hydrogel matrix in our present study. The hydrogels were prepared using a cold-stir method. The gelation temperature (T) of the hydrogels (n=3) was determined by a method of heating and inversion in a vial.
To determine the physical properties of poloxamer 407 hydrogels, we tested their storage/loss moduli, surface morphologies, and infrared emission/absorption spectra. The storage modulus, G′, and loss modulus, G″, of the hydrogels were performed on a rheometer (TA-AR-G2, State of Delaware, USA). Temperatures were confined within the range of 20°C–45°C under constant-shear strain. The shear moduli were measured at a frequency of 100 rad/s. The micro-morphologies of the dehydrated hydrogels were observed under a scanning electron microscope (SEM; Hitachi, H-7500, Tokyo, Japan).26 To accomplish this, the hydrogels were frozen and lyophilized with a vacuum freeze-dryer for 48 hours, and were then post-heated for 48 hours at 37°C. The dehydrated samples were cross-sectioned and stacked on the SEM sample plate. The samples were sputtered with gold ions (Hitachi, E-1010, Tokyo, Japan), placed in a 10 Pa vacuum, and were subsequently imaged. Fourier-transform infrared spectroscopy (FT-IR; via a Tensor II, Bruker Beijing Tech Co., Shanghai, China) was used to measure the spectra of the frozen-lyophilized samples.
In vitro protein release assay of poloxamer 407 hydrogels
To evaluate the controlled release dynamics of the poloxamer 407 hydrogels used in the present study, a transwell assay (Cat: 3450, Costar, Corning Co., NYC, USA) was conducted using 17.0% (w/w) hydrogels. FGF-21, KGF-2, or KGF-2/FGF-21 mixtures (provided by the Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, China) were separately dissolved in 17.0% (w/w) concentrations of cold poloxamer 407 solution, which were all made to a final concentration of 2 mg/mL. Then, 1 mL of poloxamer 407 protein solution was added into the upper compartment of transwell inserts with a 0.4 µm pore size, and the inserts were then incubated at 37°C for gelation. Once gelled, samples were placed in 6-well plates with 1 mL of 0.9% saline in the lower compartment. The saline solution in the lower compartment was analyzed for diffused FGF-21, KGF-2, or KGF-2/FGF-21 mixtures after 0.25, 0.5, 1, 2, 4, 6, 8, 10, and 20 hours. Diffused protein was quantified via a BCA Protein Assay Kit (TransGen Biotech Co., Beijing, China).
Bioactivity and cytotoxicity assays of poloxamer 407 hydrogels loaded with KGF-2 and FGF-21
The bioactivities of hydrogels loaded with KGF-2 and/or FGF-21 were investigated using a 3-[4,5-dimethylthiazole-2-yl]−2,5-diphenyltetrazolium bromide (MTT, Sigma, USA) assay in NIH 3T3 cells (purchased from ATCC, USA), as described in our previous study.24 Annexin V-fluorescein isothiocyanate (FITC) staining was performed to determine the toxicity of hydrogels, according to the manufacturer’s protocol (FITC annexin V apoptosis detection kit, Cat: 556547, BI, Israel). After the stain was applied, an anti-fluorescent quenching polyvinyl pyrrolidone (PVP) sealant (Beyotime, Shanghai, China) was used on the samples, which were then photographed with a laser-scanning confocal microscope (Leica, Weztlar, Germany). The mean fluorescent intensities of the samples were analyzed via Image Pro plus V.6.0. A scratch test was used to investigate the migratory effects of hydrogels in NIH 3T3 cells. A sterilized glass slide was used to gently traverse the bottom surface of a culture plate. The cells were observed with an inverted microscope (Nikon Eclipse TI-S, Tokyo, Japan), at a 100-fold magnification, at both 0 hour (baseline) and 24 hours (endpoint). Migratory distance was analyzed via Image Pro plus V.6.0. The cells were divided into eight groups (online supplementary figure 2). The control group (Con group) was incubated in starvation medium containing heparin sodium but no poloxamer 407. The hydrogel group was only exposed to poloxamer 407. The FGF-21 hydrogel group contained poloxamer 407 and 40 µg/mL of FGF-21. The KGF-2 hydrogel group contained poloxamer 407 and 25 µg/mL of KGF-2. The FGF-21 and KGF-2 (F21–K2) hydrogel group contained poloxamer 407, 40 µg/mL of FGF-21, and 25 µg/mL of KGF-2. The FGF-21 group contained 40 µg/mL of FGF-21 but no poloxamer 407. The KGF-2 group contained 25 µg/mL of KGF-2 but no poloxamer 407. Finally, the F21–K2 group contained 40 µg/mL of FGF-21 combined with 25 µg/mL of KGF-2 but no poloxamer 407.
Supplemental material
Animals
Twenty-four clean-grade Goto-Kakizaki (GK) rats (12 weeks old) were purchased from Shanghai Slack Laboratory Animals Co. All animals were adaptively provided with drinking water and solid pellets in cages at the Animal Experiments Center of Wenzhou Medical University. Room temperature was kept at approximately 23°C, with a relative humidity of 60% and a 12/12 hour light/dark cycle. All experimental rats were kept for 4 weeks, during which blood glucose levels were measured and recorded. All rats were randomly numbered and divided into two groups, with 12 rats being used for each group.
Scalded skin model and drug administration in rats
GK rats were anesthetized with 2.0% (w/v) pentobarbital via intraperitoneal injections. The back of each rat was depilated with a depilatory agent, which was then followed by the use of a YLS-5Q-type scald (Model: YLS-5Q, Yiting Technology Development Co., Jinan, China) to induce deep second-degree burns (1.0 cm in diameter each) on both sides of the spine. The scalding conditions were as follows: hot-head temperature of 85°C, force on the skin equal to a weight of 0.5 kg, and the duration of 10 s. The wounds were not covered, and each rat was then kept isolated in a single cage, with common rat food provided. Then, the various treatments, once daily, began on the day of wound formation. The wounds were properly treated with hydrogen peroxide and were disinfected with iodophor before hydrogel treatments began. Ultimately, 24 GK rats were divided into two groups. One group was administrated with a hydrogel containing 50 µg/mL of KGF-2 on one side and 0.9% saline on the other side as an internal control. The other group was given a hydrogel containing 500 µg/mL of FGF-21 on one side, whereas the other side was administered a hydrogel containing the same amount of KGF-2 and FGF-21. The treatment was continued for 31 days; on days 7, 14, 25, and 31, the wounds were photographed using a digital camera, and the wound healing process was analyzed via Image Pro plus V.6.0. The wound healing rate was calculated at each time point.24
H&E staining
Three GK rats from each group were euthanized with 2.0% (w/v) pentobarbital by intraperitoneal injection at a dose of 0.6 mL per 100 g on days 7, 14, 25, and 31. The skins of each rat were cut along the wound-healing perimeter after the rats were sacrificed via anesthesia.
Half of each tissue sample was fixed in 4% (w/v) paraformaldehyde, embedded in paraffin, sectioned (5 µm thickness), and mounted onto poly-L-lysine-coated slides for H&E staining (Beyotime, Shanghai, China), and immunohistochemistry (IHC). Sections were photographed with a upright Nikon microscope (ECLPSE 80i, Tokyo, Japan) at 40-fold or 100-fold magnification to view the wound area. The other half of each tissue sample was placed in a 1.5 mL Eppendorf tube, labeled, dehydrated overnight with a 20% (w/v) sucrose solution, embedded with optimum cutting temperature (OCT) compound embedding agent, and then stored at −80°C until later use for analysis via immunofluorescence (IF).
Immunohistochemistry
Mounted paraffin sections were placed in a 65°C incubator for 5 hours, after which they were dewaxed with xylene and hydrated with a decreasing gradient of alcohol solutions. The sections were then placed in 3% (w/v) hydrogen peroxide (diluted with 80% (w/v) methanol) for 10 min at 4°C, for fixation and elimination of endogenous enzymatic activity. To achieve antigen retrieval, 0.01 M sodium citrate buffer (pH=6.0) was heated to 100°C (mildly boiling) in an uncovered pressure cooker. Slides were then slowly placed inside the pressure cooker, which was then capped and sealed. After 2 min, the cooker was removed from the heat source and the pressure was allowed to vent. The slides were allowed to remain in the cooker at a temperature above 95°C for 15 min. Then, the lid was opened, the contents were allowed to cool, and the slides were washed three times (5 min per wash) with phosphate buffer saline (PBS) to remove the sodium citrate. Next, the slides were placed in a wet box, blocked with 5% (w/v) goat serum (Solarbio, Shanghai, China, diluted with 0.01 M PBS) for 1 hour in a 37°C incubator, and then incubated with specific primary antibodies (each diluted in 1% (w/v) goat serum) at 4°C overnight. The primary antibodies included the following: collagen III (22734-1-AP, 1:250, Proteintech, Wuhan, China), transforming growth factor-β (TGF-β) (21898-1-AP, 1:250, Proteintech), vascular endothelial growth factor (VEGF) (19003-1-AP, 1:250, Proteintech), interleukin (IL)-6 (21865-1-AP, 1:100, Proteintech), IL-10 (60269-1-Ig, 1:100, Proteintech), and KGF-2 (also named FGF-10 antibody, #32224, 1:200, Signalway, Maryland, USA). Subsequently, samples were incubated in goat anti-rabbit IgG-HRP or goat anti-mouse IgG-HRP (1:100, TransGen Biotech Co., Beijing, China) for 1 hour in a 37°C incubator. The sections were then washed four times with PBS (5 min per wash) and stained with 3,3N-diaminobenzidine tertrahydrochloride. Subsequently, sections were stained with hematoxylin for 5 min, differentiated with 0.5% (w/v) hydrochloric acid for 5 s, dehydrated, and sealed with neutral resin to prevent air bubbles. Three random areas were photographed from each section at 400-fold magnification, using a Nikon ECLPSE 80i microscope (Nikon, Tokyo, Japan). The mean immunohistochemical-staining intensity was calculated via Image Pro Plus V.6.0.
Immunofluorescence
Tissue embedded in OCT was frozen, sliced to a thickness of 10 µm, and stored at −20°C until further use. Then, the frozen slides were placed at room temperature for 10 min, washed four times with PBS (5 min per wash), and blocked with 5% (w/v) goat serum for 1 hour at 37°C. The excess goat serum was aspirated, and the primary antibodies were incubated at 4°C overnight. The primary antibodies included the following: alpha-smooth muscle actin (α-SMA) (55135-1-AP, 1:250, Proteintech), Pan-Keratin (4545S, 1:250, CST, Danvers, MA, USA), and cluster of differentiation 31, CD31 (40699-1, 1:200, SAB), all of which were diluted with 1% (w/v) goat serum. The next day, the sections were rewarmed for 40 min at room temperature, washed four times with PBS (5 min per wash), and then incubated with specific secondary antibodies (goat anti-rabbit IgG (H+L)-AF488, 1:200, TransGen biotech Co.; goat anti-mouse IgG (H+L)-AF488, 1:250, Gibco, all diluted with 1% (w/v) goat serum) for 1 hour in the dark at 37°C. The sections were washed four times with PBS (5 min per wash) and stained with 4',6-diamidino-2-phenylindole (DAPI) for 15 min in the dark. After four subsequent washes in PBS (5 min per wash), the anti-fluorescent quencher PVP (Beyotime, Shanghai, China) was added to the tissue to seal the slides. Three random areas of each section were photographed at a 400-fold magnification using a Leica laser confocal microscope (Leica, Germany). The mean fluorescent intensity of each image was analyzed via Image Pro Plus V.6.0.
Western blotting
Proteins from animals or cells were quantified via BCA reagents (Beyotime, China), and equivalent amounts of protein (40 µg in vitro). The extracted protein was mixed with loading buffer and boiled and stored at −20°C. In addition, 20 µL of protein mixtures containing 40 µg of total proteins were loaded onto 12% polyacrylamide gels and were electrophoretically separated at 80 V. After 2 hours, the separated protein was blotted onto a polyvinylidene fluoride (PVDF) membrane at an electric current of 300 mA for 90 min. Subsequently, the PVDF membrane was blocked with 5% skimmed milk (BD/DicoTM, State of New Jersey, USA) in tris-buffered saline tween-20 (TBST) for 90 min. Subsequently, corresponding primary antibodies and probes were added and samples were incubated at 4°C overnight. The primary antibodies included the following: α-SMA (55135-1-AP, 1:1000, Proteintech), Collagen III (22734-1-AP, 1:800, Proteintech), Pan-Keratin (4545S, 1:1000, CST), VEGF (19003-1-AP, 1:1000, Proteintech), CD31 (40699-1, 1:1000, SAB), TGF-β (21898-1-AP, 1:800, Proteintech), IL-6 (21865-1-AP, 1:1000, Proteintech), IL-1β (66737-1-Ig, 1:2000, Proteintech), tumor necrosis factor-α (TNF-α) (66737-2-Ig, 1:2000, Proteintech), IL-10 (60269-1-Ig, 1:4000, Proteintech), and KGF-2 (#32224, 1:1000, Signalway). Next, samples were incubated with the corresponding secondary antibodies (goat IgG, anti-mouse IgG, or goat anti-rabbit IgG (1:5000) conjugated with horseradish peroxidase (HRP)) for 1 hour at room temperature. Subsequently, the PVDF membrane was washed three times with TBST, after which signals were detected by an electrochemiluminescence (ECL) chemiluminescent agent, and the results were captured via a ChemiDoc XRS+Imaging System (Bio-Rad). Finally, the grey values of the target bands were analyzed by the Image lab software.
Statistical analysis
Graphpad Prism V.6.0 was used for all statistical analysis. Data are expressed as the mean±SD. Comparisons between two groups were performed using t tests. Comparisons among three or more groups were performed using analyses of variance. Statistical significance was considered when p<0.05.
Results
KGF-2/FGF-21 poloxamer hydrogels exhibit thermosensitive characteristics with significant sustained-release properties
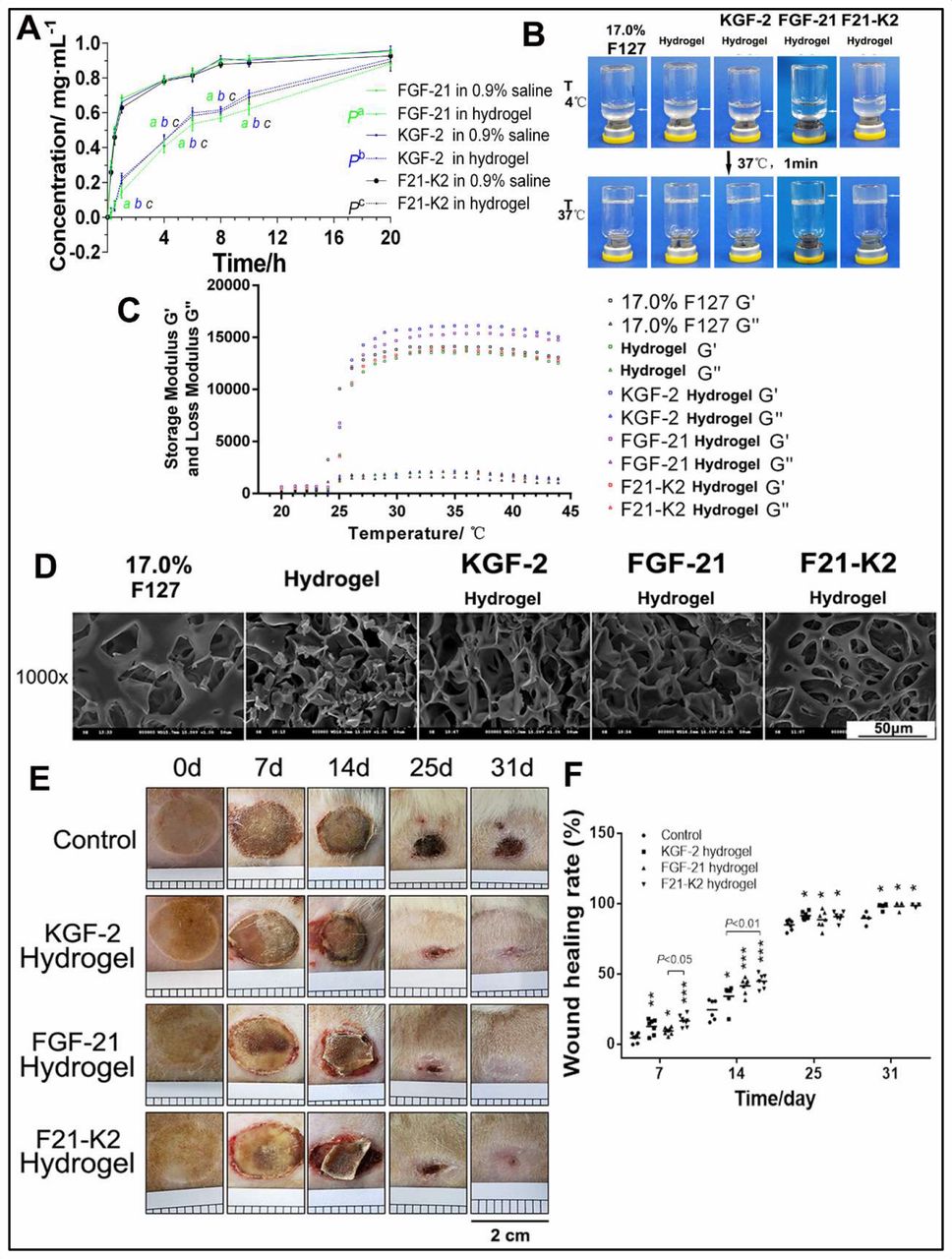
Poloxamer 407 is a typical thermosensitive and viscoelastic material. As shown in figure 1C, the optimized poloxamer 407 hydrogel formulation revealed a sol-gel transition at 37°C. Except for the gelation temperature, both the storage modulus (G′) and loss modulus (G″) were important viscoelastic parameters. The storage modulus and loss modulus of the poloxamer 407 hydrogel, as well as the KGF-2-loaded and FGF-21-loaded hydrogels, were also characterized (figure 1C). The similarities in the ratios of storage-to-loss moduli between these hydrogels indicated that they were at a similarly solid state. However, due to the use of glycerol and proteins, the storage modulus (G′) and loss modulus (G″) of drug-loaded hydrogels increased, which concomitantly improved elasticity. Furthermore, the structures of poloxamer 407 hydrogels and KGF-2-loaded and/or FGF-21-loaded hydrogels were observed via SEM. Holes were found on the surfaces and on the insides of the poloxamer 407 hydrogels, as well as in KGF-2-loaded and/or FGF-21-loaded hydrogels, with interconnected, sponge-like structures (figure 1D). Taken together, the SEM images of KGF-2-loaded and/or FGF-21-loaded hydrogels indicated that their three-dimensional (3D) structures might be conducive to growth-factor incorporation and controlled release. FT-IR spectroscopy results showed similar spectral signatures in each hydrogel group (online supplementary figure 1), which confirmed the presence of integrated KGF-2 and/or FGF-21 proteins.

Characteristic of thermosensitive hydrogel. (A) Control release profile of FGF-21, KGF-2 and KGF-2/FGF-21 mixtures loaded hydrogel compared with that of corresponding protein in 0.9% saline respectively (n=3). ‘a, b, c’ indicated as significantly different from corresponding protein in 0.9% saline respectively (p<0.01). (B) Gelation of thermosensitive hydrogel. (C) Storage modulus and loss modulus of thermosensitive hydrogel. (D) SEM images of thermosensitive hydrogel (scale bars, 50 µm). (E) Skin wound of GK rat (scales 0.2 cm per square). (F) Wound healing rate. compared with the control group, *p<0.05, **p<0.01, ***p<0.001 (n=7 on day 7, n=6 on day 14, n=6 on day 25, n=3 on day 31). FGF-21, fibroblast growth factor-21; F21–K2, FGF-21 and KGF-2; GK, Goto-Kakizaki; KGF-2, keratinocyte growth factor-2; SEM, scanning electron microscope.
We next compared the in vitro release profiles of 17.0% poloxamer 407 hydrogels with FGF-21, KGF-2, or KGF-2/FGF-21 mixtures. As shown in figure 1A, slow release of FGF-21, KGF-2, or KGF-2/FGF-21 mixtures were observed for poloxamer 407 and was significantly different from that of corresponding protein in 0.9% saline (pa<0.01, pb <0.01, pc <0.01). After 20 hours, complete release was achieved in three poloxamer 407 hydrogel protein groups. However, there was no significant difference in the in vitro release profiles among the three poloxamer 407 hydrogel protein groups (p>0.05).
Bioactivities and cytotoxicities of poloxamer 407 hydrogels loaded with KGF-2 and FGF-21
We next investigated the bioactivities and cytotoxicities of hydrogels in terms of in vitro cellular proliferation, apoptosis, and migration to determine the biosafety of poloxamer 407 in such preparations. As shown in online supplementary figure 2, MTT cellular proliferation data (online supplementary figure 2A) suggested that poloxamer 407 hydrogels did not inhibit cellular bioactivity (p>0.05) when compared with that of the control group. Similarly, compared with that of the control group, FGF-21-loaded hydrogels did not inhibit cellular proliferation. In the F21–K2 hydrogel group, cellular bioactivity appeared to be slowed down but was not statistically significant compared with that of the control group (p>0.05). In the apoptotic experiment, we found that the poloxamer 407 hydrogel alone induced significant apoptosis (p<0.01) and high expression of Annexin-V (online supplementary figure 2D,E). However, once the hydrogel was loaded with KGF-2 and/or FGF-21, due to the protective effects of these growth factors, apoptosis was effectively prevented (p>0.05). Moreover, in the cellular migration experiments (online supplementary figure 2B,C), compared with that of the control group, poloxamer 407 hydrogels alone did not promote cellular migration, while KGF-2 or FGF-21 hydrogels each enhanced cellular migration (both p<0.01). The F21–K2 hydrogel group also significantly enhanced cellular migration (both p<0.01). Furthermore, the F21–K2 hydrogel group demonstrated the strongest enhancement in cellular migration compared with that of the other groups (p<0.05).
KGF-2/FGF-21 poloxamer hydrogels accelerate wound closure and promote re-epithelialization in rats
To evaluate whether the combination of KGF-2 and FGF-21 in poloxamer hydrogels improve diabetic wound healing in comparison with that in the saline vehicle group, scalded skin wounds were administered in GK rats. Figure 1E shows a representative series of scalded wounds on days 0, 7, 14, 25, and 31 during the four different treatments. As depicted in figure 1F, when compared with that of the control group, all other treatment groups (KGF-2 hydrogel, FGF-21 hydrogel, and F21–K2 hydrogel) demonstrated an improvement in the wound healing rate of scalded skin in GK rats on days 7, 14, 25, and 31. Among the three groups, the F21–K2 hydrogel group exhibited the best overall improvement. On day 7, the F21–K2 hydrogel group showed a higher wound healing rate compared with that of the FGF-21 hydrogel group (p<0.05). On day 14, the F21–K2 hydrogel group showed a higher wound healing rate than that of the KGF-2 hydrogel group (p<0.01). On days 25 and 31, there was no significant difference in the healing rates of the three treatment groups, but each of these healing rates were still higher than that of the control group. In addition, to evaluate re-epithelialization in the F21–K2 hydrogel group, H&E staining of epidermal thickness on day 31 was evaluated. As shown in figure 2A,B, the epidermal thickness in the KGF-2 hydrogel and F21–K2 hydrogel groups was much thicker than that of the control group on day 31 (p<0.01).

Results of H&E staining and expression of α-SMA and collagen III measured with immunostaining and Western blot. (A) Epidermis by H&E staining on day 31 (red arrow indicates epiderm, yellow line indicates epidermis thickness, scale bar: 250 µm). (B) Statistical analysis of epidermis thickness (n=3). (C) H&E staining of full thickness of skin on day 31 (yellow arrow indicates bubble, blue arrow indicates hair follicle, left magnification is 40 folds; right magnification is 100 folds, scale bar: 250 µm). (D) IF of α-SMA in dermis (scale bar: 25 µm). (E) Statistical analysis of α-SMA expression by IF in dermis (n=3). (F) Western blot of α-SMA in wound section on day 25 and 31. (G) Statistical analysis of α-SMA expression by Western blot. (H) Immunostaining of collagen III in dermis on day 25 and 31 (scale bar: 25 µm). (I) Statistical analysis of collagen III expression by IHC in dermis (n=3). (J) Western blot of collagen III in wound section on day 25 and 31. (K) Statistical analysis of collagen III expression by Western blot. Compared with the control group, *p<0.05, **p<0.01 (n=3). DAPI, 4',6-diamidino-2-phenylindole; FGF-21, fibroblast growth factor-21; F21–K2, FGF-21 and KGF-2; IF, immunofluorescence; IHC, immunohistochemistry; KGF-2, keratinocyte growth factor-2; α-SMA, alpha-smooth muscle actin.
KGF-2/FGF-21 poloxamer hydrogels enhance granulation formation via increased α-SMA expression
Skin tissue from each group on day 31 was used for histologic analysis. H&E staining confirmed the superiority of the F21–K2 hydrogel group in terms of its histopathological repair (figure 2C). The epithelium was intact in the F21–K2 hydrogel group, and the collagen in the dermis layer was neatly arranged without any obvious edema. Meanwhile, its peripheral region was well integrated into the surrounding native skin tissue, and the formation of many skin appendages and hair follicles (blue arrow in figure 2C) was observed. In contrast, the dermal layer was still collapsed in the FGF-21 hydrogel group (red-dotted frame in figure 2C), and dermal edema (yellow arrow in figure 2C) was still present in the control group, with no obvious skin appendage regeneration being observed.
IF staining showed that the expression levels of α-SMA in the dermis in the KGF-2 hydrogel, FGF-21 hydrogel, and F21–K2 hydrogel groups were significantly increased on day 31, as compared with those of the control group (p<0.001, p<0.05, p<0.001, figure 2D,E). Moreover, compared with that of the FGF-21 hydrogel group, the expression levels of α-SMA in the KGF-2 hydrogel group and F21–K2 hydrogel group were significantly higher (p<0.01). Western blotting also showed significantly increased expression levels of α-SMA in the three treatment groups on days 25 and 31 (figure 2F,G).
KGF-2/FGF-21 poloxamer hydrogels increase collagen synthesis
Since collagen III plays an essential role in new capillary formation, immunohistochemical staining and Western blotting were used to evaluate collagen-III protein levels. As shown in figure 2H,I a significantly increased protein level of collagen III was found in the KGF-2 hydrogel group and the F21–K2 hydrogel group on day 25, as compared with those of the control group (p<0.01). Moreover, the expression levels of collagen III were significantly increased in all three treatment groups on day 31 (p<0.001, p<0.01, p<0.001), as compared with that of the control group. Meanwhile, the F21–K2 hydrogel group exhibited a significantly higher level of collagen III compared with those of the other treatment groups (p<0.01). As shown in figure 2J,K, Western blotting similarly revealed significantly increased protein levels of collagen III in the three treatment groups in whole-skin-wound sections on day 31, as compared with that of the control group (p<0.01).
KGF-2/FGF-21 poloxamer hydrogels promote re-epithelialization and proliferation via increased pan-keratin, TGF-β and KGF-2 levels
Pan-keratin is a marker of re-epithelialization of the extracellular matrix. Immunohistochemical staining (figure 3A,B) showed higher pan-keratin levels in the three treatment groups on day 31 (p<0.01, p<0.05, p<0.001), as compared with that of the control group. Specifically, the F21–K2 hydrogel group showed the strongest effect. Western-blot results (figure 3C,D) showed similar protein levels of pan-keratin in the three treatment groups on days 7 and 31 (p<0.05).
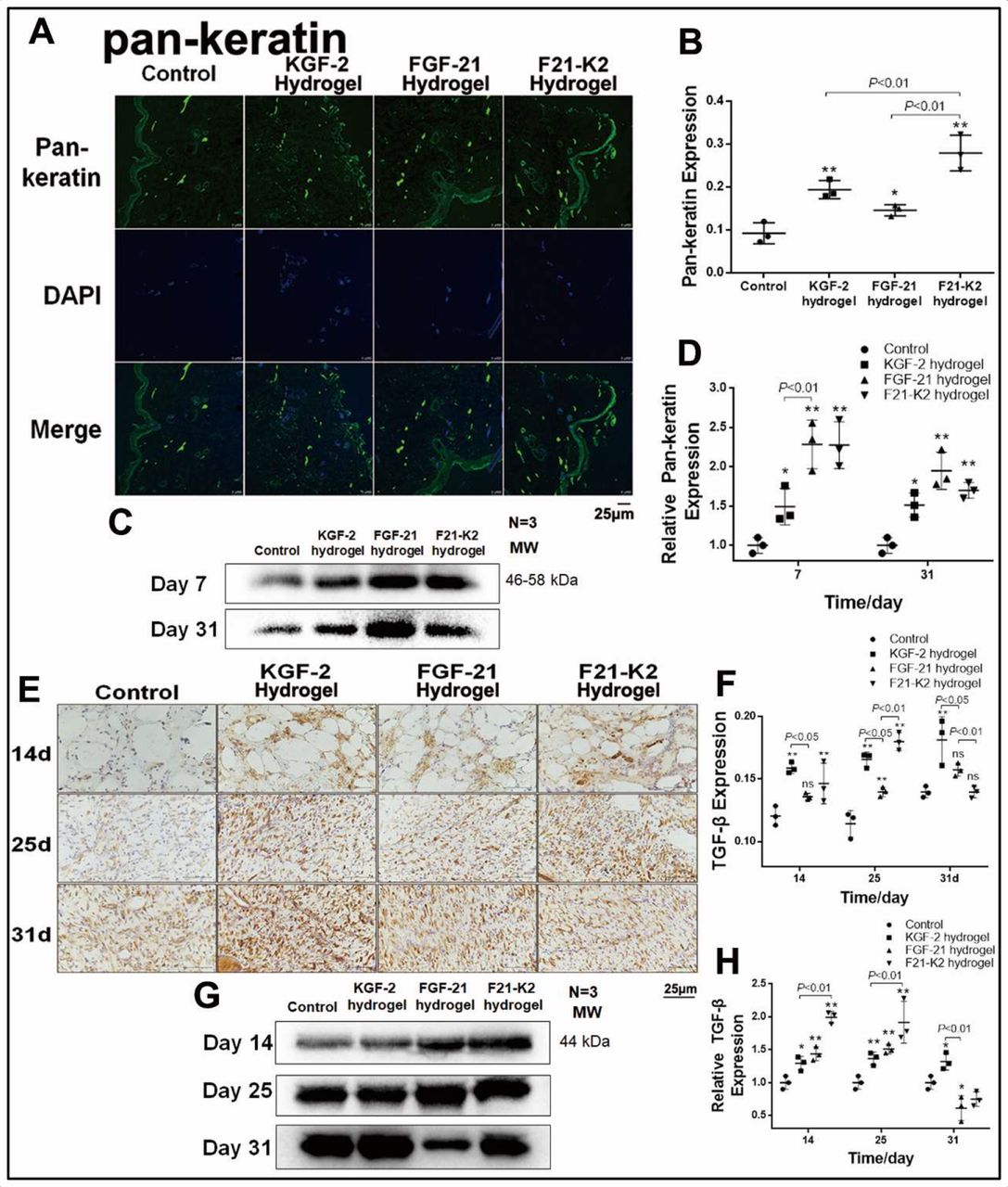
Results of promoted proliferation. (A) IF of pan-keratin in dermis (green object indicates pan-keratin positive, scale bar: 25 µm). (B) Statistical analysis of pan-keratin expression by IF in dermis (n=3). (C) Western blot of pan-keratin in wound section on days 7 and 31. (D) Statistical analysis of pan-keratin expression measured with Western blot (n=3). (E) Immunostaining of TGF-β in dermis on days 14, 25 and 31 (brown object indicates pan-keratin positive, scale bar: 25 µm). (F) Statistical analysis of TGF-β expression measured with IHC in dermis (n=3). (G) Western blot of TGF-β in wound section on days 14, 25 and 31. (H) Statistical analysis of TGF-β expression measured with Western blot (n=3). Compared with the control group, *p<0.05, **p<0.01. DAPI, 4',6-diamidino-2-phenylindole; FGF-21, fibroblast growth factor-21; F21–K2, FGF-21 and KGF-2; IF, immunofluorescence; KGF-2, keratinocyte growth factor-2; TGF-β, transforming growth factor-β.
To evaluate proliferative effects, the TGF-β expression was evaluated on days 7, 25, and 31. Immunohistochemical staining demonstrated that the KGF-2 and F21–K2 hydrogel groups showed significantly higher TGF-β expression compared with that of the control group on days 14 and 25 (p<0.001, p<0.01, respectively, figure 3E,F). The FGF-21 hydrogel group only showed a slight increase of TGF-β on day 14 (p<0.05). Western-blot results (figure 3G,H) also revealed higher TGF-β levels in the three treatment groups on days 14 and 25 (p<0.05), as compared with that of the control group. Similarly, the F21–K2 hydrogel exhibited the highest expression of TGF-β on days 14 and 25, as compared with those of the other treatment groups (figure 3G,H, p<0.01).
Next, we detected expression levels of KGF-2 in skin-wound sections via immunohistochemical staining and Western blotting. As shown in online supplementary figure 3, the expression level of KGF-2 increased gradually on days 7, 25, and 31, which indicated that the reduction of the wound area in the control group was correlated with an increase in endogenous KGF-2 expression. In addition, the FGF-21 group also confirmed that the expression of KGF-2 was up-regulated at the early stage (on day 7) of wound healing and that endogenous expression of KGF-2 was significantly down-regulated as the wound was nearly completely healed on day 31.
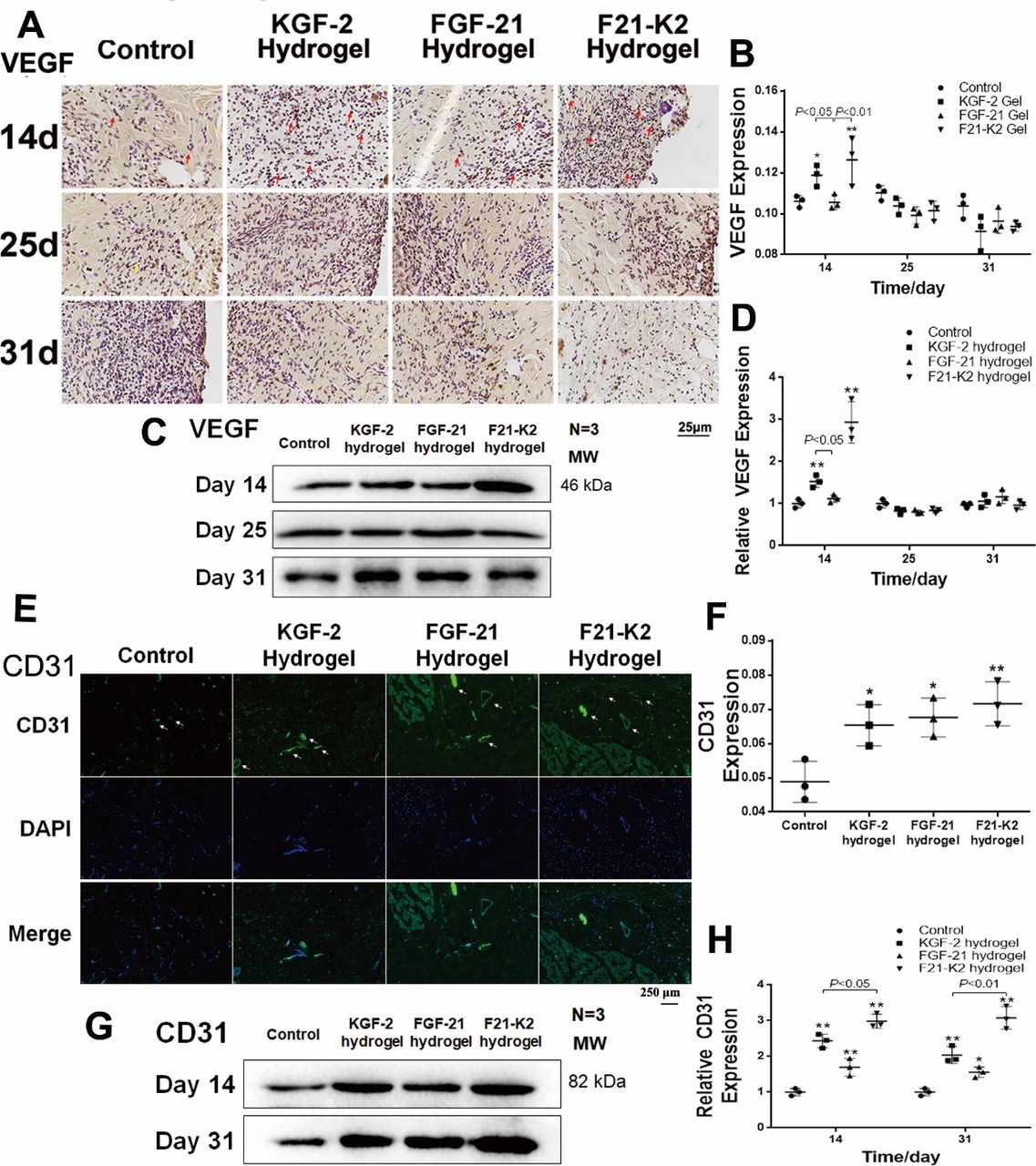
KGF-2/FGF-21 poloxamer hydrogels increase angiogenesis via increasing VEGF and CD31 levels
Proper wound healing requires angiogenesis in the newly generated dermis.33 On days 14, 25, and 31, skin tissues were assessed via immunohistochemical staining and Western blotting to quantify the expression of VEGF, which is a biomarker of endothelial cells in blood vessels. As shown in figure 4A–D, the expression of VEGF was significantly increased in both KGF-2 hydrogel and F21–K2 hydrogel groups on day 14 (p<0.05).
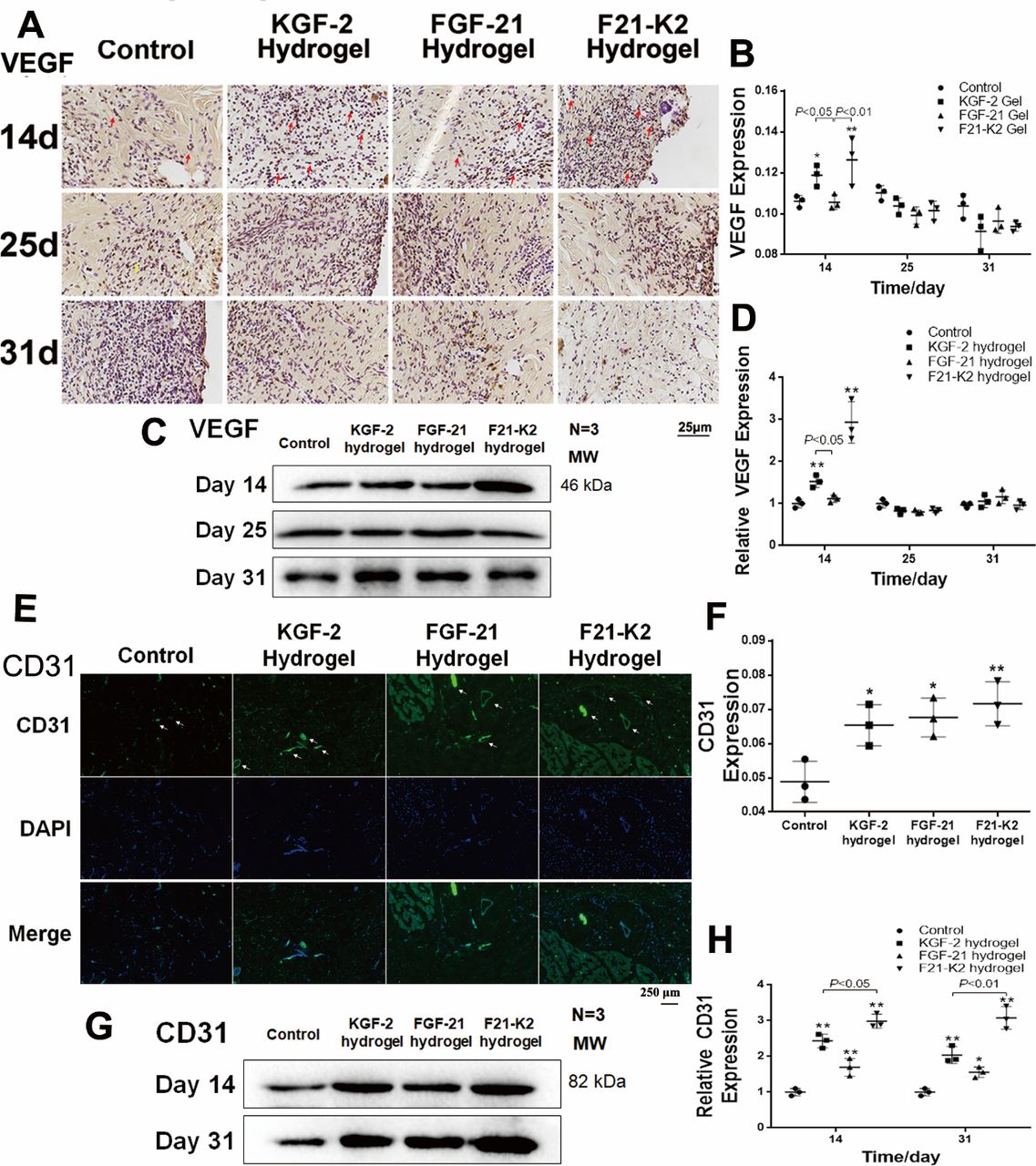
Protein expression of VEGF and CD31 measured with immunostaining and Western blot. (A) Immunostaining of VEGF in dermis (red arrow indicates positive object, bar: 25 µm). (B) Statistical analysis of VEGF measured with IHC in dermis (n=3). (C) Western blot of VEGF in wound section on days 14, 25 and 31. (D) Statistical analysis of VEGF expression measured with Western blot (n=3). (E) IF of CD31 in dermis on day 31 (white arrow indicates blood vessel, scale bar: 250 µm). (F) Statistical analysis of CD31 expression measured with IF in dermis (n=3). (G) Western blot of CD31 in wound section on days 14 and 31. (H) Statistical analysis of CD31 expression measured with Western blot (n=3). Compared with the control group, *p<0.05, **p<0.01, (n=3). DAPI, 4',6-diamidino-2-phenylindole; FGF-21, fibroblast growth factor-21; F21–K2, FGF-21 and KGF-2; IF, immunofluorescence; KGF-2, keratinocyte growth factor-2; VEGF, vascular endothelial growth factor.
In addition, on day 14 or 31, skin tissue sections assessed via immunofluorescence and Western blotting to quantify the expression of CD31, another biomarker of endothelial cells in blood vessels. As shown in figure 4E alarge number of newly formed vessels (white arrows, figure 4E) were observed at the wound bed in KGF-2 hydrogel, FGF-21 hydrogel, and F21–K2 hydrogel groups. However, in the control group, only a few endothelial cells with CD31 expression were found. Western-blot results (figure 4G,H) showed higher CD31 levels in the three treatment groups on days 14 and 31 (p<0.05), as compared with that of the control group. Similarly, the CD31 levels (figure 4H) indicated that the F21–K2 hydrogel had the highest blood vessel density, as compared with that of the FGF-21 hydrogel alone (p<0.05).
KGF-2/FGF-21 poloxamer hydrogels play an important role in the regulation of inflammatory cytokines
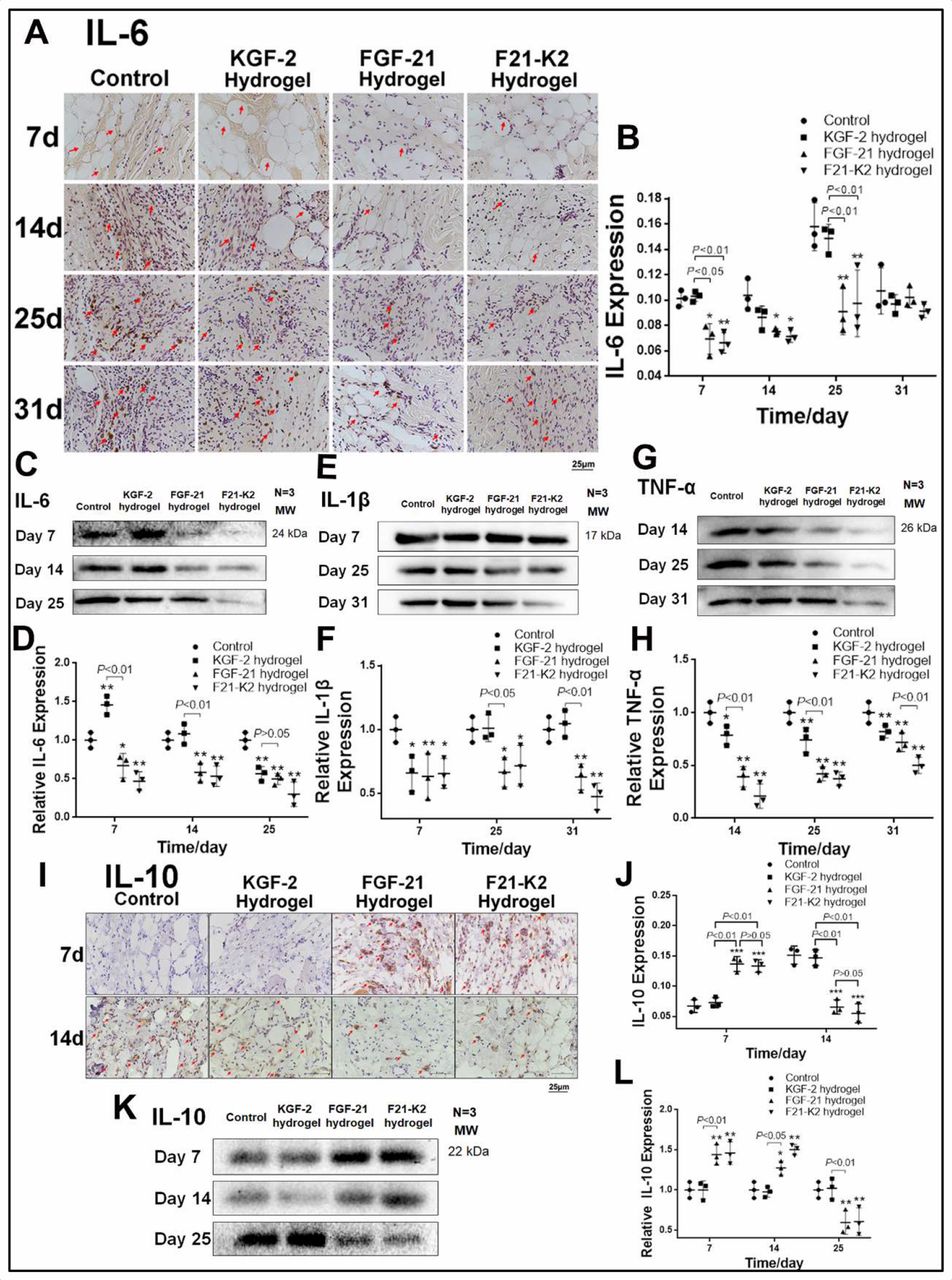
Inflammation is an essential process during wound healing. To investigate whether KGF-2/FGF-21 poloxamer 407 hydrogels regulate inflammation in wounded tissue, we measured the expression levels of pro-inflammatory factors (IL-6, IL-1β, and TNF-α) and an anti-inflammatory factor (IL-10).
As shown in figure 5A–D, on days 7, 14, and 25, immunohistochemical staining and Western blotting showed that the expression of IL-6 was significantly decreased in the FGF-21 and F21–K2 hydrogel groups compared with that in the control group (p<0.05, p<0.01, respectively). Moreover, the expression of IL-1β in Western blotting (figure 5E,F) was significantly decreased in the FGF-21 and F21–K2 hydrogel groups on days 14 and 25 (p<0.05), as compared with that of the control group. In addition, the expression of TNF-α (figure 5G,H) was significantly decreased on days 14, 25, and 31 (p<0.05) in all three treatment groups compared with that in the control group. However, the FGF-21 and F21–K2 hydrogel groups had significantly decreased expression of TNF-α compared with that of the KGF-2 group (p<0.01).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of pro-inflammatory factors IL-6, IL-1β, TNF-α and anti-inflammatory factor IL-10. (A) Immunostaining of IL-6 in dermis (red arrow indicates IL-6 positive object, scale bar: 25 µm). (B) Statistical analysis of IL-6 expression measured with IHC in dermis (n=3). (C, E, G) Western blot of IL-6, IL-1β and TNF-α in wound section, respectively. (D, F, H) Statistical analysis of IL-6, IL-1β and TNF-α expression measured with Western blot, respectively. (I) Immunostaining of IL-10 in dermis on days 7 and 14 (red arrow indicates IL-10 positive object, scale bar: 25 µm). (J) Statistical analysis of IL-10 expression by IHC in dermis (n=3). (K) Western blot of IL-10 in wound section on days 7, 14 and 25. (L) Statistical analysis of IL-10 expression measured with Western blot (n=3). Compared with the control group, *p<0.05, **p<0.01. FGF-21, fibroblast growth factor-21; F21–K2, FGF-21 and KGF-2; IL, interleukin; KGF-2, keratinocyte growth factor-2; TNF-α, Tumor necrosis factor alpha.
Immunohistochemical staining and Western blotting of IL-10, an anti-inflammatory factor, are shown in figure 5I–L. The expression of IL-10 was significantly higher on day 7 (p<0.001) and was decreased gradually on days 14 and 25 in the FGF-21 and F21–K2 hydrogel groups (p<0.01 or p<0.05), as compared with corresponding values in the control group.
Discussion
In the present study, a poloxamer 407 thermosensitive hydrogel loaded with KGF-2 and/or FGF-21 was prepared and its physical and biological properties were characterized. The repairing effects of this hydrogel were investigated in a model of scalded skin in GK rats.
Our goal was to extend the retention time of growth factors on the skin surface by virtue of the good plasticity and adhesion of the hydrogel. In order to study the plasticities of hydrogels, we measured the elastic moduli of a blank hydrogel and a drug-loaded hydrogel. As shown in figure 1C, loading with KGF-2 and FGF-21 increased the elastic moduli of poloxamer 407 hydrogels, rendering better plasticity. This result suggested that poloxamers have good compatibility with KGF-2 and FGF-21. We next examined the microstructure of KGF-2 and FGF-21 hydrogels via SEM. We found that poloxamer hydrogels exhibited a favorable 3D structure, such that the 3D web-like structure not only facilitated the slow release of KGF-2 and FGF-21 proteins at the wound site, but also may have provided an effective scaffold for cellular growth. The advantages of these 3D-scaffold structures in cellular growth and migration were demonstrated in our cell-migration assays (online supplementary figure 2).
Although poloxamer 407 is a polymer material approved by the Food and Drug Administration (FDA) for injections, high concentrations of poloxamer 407 may still be cytotoxic according to previous studies.34 Therefore, we investigated the cytotoxicity of poloxamer 407 loaded with KGF-2 and FGF-21 using three in vitro assays assessing cellular proliferation, apoptosis, and migration (online supplementary figure 2). We found that poloxamer 407 exhibited weak cytotoxicity. However, once loaded with KGF-2 and/or FGF-21, due to the protective effects of these growth factors, the cytotoxicity of the poloxamer 407 hydrogel was reduced. In addition, as shown in online supplementary figure 2B,C, hydrogels loaded with these growth factors exhibited better cell-migration effects than the use of either growth factor alone.
Next, we investigated the repairing effects of KGF-2 and FGF-21 hydrogels in a model of scalded skin in GK rats. We found that the combination of KGF-2 and FGF-21 in hydrogels exhibited a better effect in promoting wound healing than the use of a KGF-2 or FGF-21 hydrogel alone. Moreover, the F21–K2 hydrogel group showed better re-epithelialization and granulation formation compared with these parameters in the KGF-2 or FGF-21 hydrogel group alone (figure 2A,C). We found that this KGF-2/FGF-21 synergy may be attributed to KGF-2-mediated cellular proliferation and FGF-21-mediated regulation of inflammatory responses. TGF-β is a growth factor that plays an important role in the regulation of injury repair through influencing the accumulation of the extracellular matrix during the healing process. It has been reported that the expression levels of cytokines, such as TGF-β, are down-regulated in diabetic ulcer tissue, which affects the synthesis of collagen and the extracellular matrix, leading to delayed healing.35–37 As shown in figures 2 and 3, the KGF-2 hydrogel group had significantly increased expression levels of TGF-β, α-SMA, pan-keratin, and collagen III compared with these levels in the control group, which confirmed the advantages of KGF-2 in promoting re-epithelialization and granulation formation. KGF-2 also promoted angiogenesis by promoting the expression of CD31 and VEGF (figure 4). In contrast, the FGF-21 hydrogel group showed a stronger regulation of inflammatory responses. As shown in figure 5, on days 7 and 14, FGF-21 significantly decreased IL-6, IL-1β, and TNF-α expression levels, while upregulating the expression of the anti-inflammatory cytokine, IL-10, as compared with these levels in the control group. In diabetic ulcer tissue, an increased expression of pro-inflammatory cytokines, accompanied by a decrease in the expression of anti-inflammatory cytokines, usually results in a loss or delayed response of inflammatory signals in local ulcer tissues38–40; this phenomenon is considered to represent one of the primary reasons for the non-healing properties of diabetic ulcers. Diabetic ulcers are chronic wounds that are the result of repetitive trauma in the insensate foot. The presence of infection and peripheral vascular disease makes the treatment of these wounds demanding and requires a multidisciplinary approach. Our present results suggest that FGF-21 reduced excessive inflammatory responses in a model of scalded skin in rats, while accelerating the conversion of scalded skin from the inflammatory phase to the proliferative phase through upregulating IL-10 and downregulating IL-6, IL-1β, and TNF-α.
Additionally, our present study revealed that the combination of KGF-2 and FGF-21 was superior to KGF-2 or FGF-21 alone in inducing cellular proliferation and inhibiting inflammation, respectively. Moreover, we found that KGF-2 exhibited better cellular proliferation than FGF-21, meanwhile, FGF-21 had better anti-inflammatory effects than KGF-2. Therefore, the combined administration of KGF-21-KGF-2 showed a synergistic effect in anti-inflammatory and promoting cellular proliferation. This may be the main reason why the combined treatment of KGF-2 and FGF-21 has a better effect on wound repair. Hence, these cellular-proliferation and inflammation-reducing factors likely underlie the molecular mechanisms by which KGF-2/FGF-21 hydrogels promote the repair of scalded skin in rats.
Conclusion
Here, we reported the design of a KGF-2/FGF-21 poloxamer 407 hydrogel. We identified that a 17.0% (w/w) poloxamer 407 in combination with 1.0% (w/w) glycerol exhibited good release kinetics in vitro. Experiments in rats showed that KGF-2 and FGF-21 hydrogels accelerated healing of scalded skin, likely via a synergistic effect of KGF-2-mediated cellular proliferation and FGF-21-mediated inhibition of inflammation.
Acknowledgments
The authors would like to thank proteins provided by the Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou, China.
References
Footnotes
Contributors XW, XL, and XY conceived and designed the experiments. XY, QZ, RY, YZ, MC, and CL performed the experiments. QH analyzed and constructed the data figures. JB, WS, and TH contributed to reagents/analysis tools. LL and JG contributed materials. XW and XY wrote the manuscript. XW is the guarantor of this work and, as such, had full access to all the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis.
Funding This work was supported by a grant from the Ministry of Science and Technology of China (No. 2016ZX09101117), the National Natural Science Foundation of China (No. 81601695), and the Natural Science Foundation of the Zhejiang Province (No. LY17H150002 and No. LY17H300003).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All animal experiments were handled in accordance with the IACUC guidelines of Wenzhou Medical University (Zhejiang, China), which comply with NIH guidelines for the care and use of laboratory animals. All animal experiments were approved by the Animal Institutional Ethics Committee of Wenzhou Medical University (No. wydw2017-0102, Wenzhou, Zhejiang, China) and were performed following the guidelines for animal experimentation at Wenzhou Medical University. Humane treatment of all research animals was assured. This article does not contain any studies with human participants. All authors confirm that ethical principles were followed in the experiments of the present study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.