Article Text

Abstract
Objective Diabetic cardiomyopathy (DCM) is characterized by cardiac remodeling, dysfunction, and insulin resistance; however, the underlying mechanism has not been fully elucidated. Programmed cell death 4 (PDCD4) is a novel inflammation and apoptosis gene, but its role in type 2 DCM remains elusive. We aimed to determine if PDCD4 intervention improves DCM by affecting left ventricular remodeling, function, and insulin resistance.
Research design and methods We designed a PDCD4-/- rat, established a type 2 diabetes animal model, and constructed a PDCD4 overexpressed adenovirus and PDCD4 small interfer RNA (siRNA) vectors to alter PDCD4 expression in H9c2 cardiomyocytes. Thereafter, glucose levels, lipid metabolism, echocardiography, and extent of myocardial fibrosis, inflammation, and apoptosis were compared in vivo and in vitro.
Results PDCD4 deficiency improved insulin resistance, cardiac remodeling, and dysfunction in type 2 DCM rats and improved myocardial hypertrophy, fibrosis, inflammation, and apoptosis. Proliferation and transformation of cardiac fibroblasts was reduced via PDCD4 downregulation in vitro under high-glucose stimulation. Furthermore, PDCD4 regulated the myocardial phosphatidylinositol 3-kinase-protein kinase B (PI3K-AKT) pathway in vivo and in vitro. PDCD4 intervention affected cardiac remodeling, dysfunction, and insulin resistance by influencing fibrosis, inflammation, and apoptosis via the PI3K-AKT pathway in vivo.
Conclusions PDCD4 knockdown protected against left ventricular remodeling, dysfunction, and insulin resistance in type 2 DCM rats. The underlying mechanisms may involve reducing cardiomyocyte apoptosis, inflammation, fibrosis, and normalized PI3K-AKT phosphorylation. To the best of our knowledge, our study is the first to report the effects and underlying mechanisms of PDCD4 in type 2 DCM. These results provide a potential new treatment avenue for improving the prognosis of patients with type 2 DCM.
- cardiac diabetic complication
- cell and molecular biology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Programmed cell death 4 (PDCD4) is a novel inflammation and apoptosis gene, but its role in type 2 diabetic cardiomyopathy (DCM) remains elusive.
What are the new findings?
PDCD4 deficiency improved insulin resistance, cardiac remodeling, and dysfunction in type 2 DCM rats and improved myocardial hypertrophy, fibrosis, inflammation, and apoptosis via the PI3K-AKT pathway in vivo.
Under high-glucose stimulation, PDCD4 downregulation reduced proliferation and transformation of cardiac fibroblasts in vitro.
The PI3K-AKT pathway is closely related to PDCD4’s involvement in the regulation of type 2 DCM.
How might these results change the focus of research or clinical practice?
PDCD4 intervention may provide an important treatment target for DCM and broaden new exploration ideas to improve DCM prognosis. Further studies with more experiments are carried out to determine other possible functional mechanisms of PDCD4 with type 2 DCM; for example, PDCD4 expression could be influenced through miRNA 21 and miR-499 activities.
Introduction
Diabetes has long been a threat to global health, and cardiovascular disease is the leading cause of heart failure and death in type 2 diabetics.1–3 It is characterized by left ventricular (LV) remodeling, diastolic dysfunction, and sustained hyperglycemia.2 4–6 Meanwhile, insulin resistance,7 8 oxidative stress,2 7 and myocardial metabolic disturbances5 are involved in this process. In addition, fibrosis,9 inflammation,10–12 and apoptosis5 are also involved in the development of diabetic cardiomyopathy (DCM). However, the pathogenic mechanism of DCM remains unclear.
A series of inflammation-related genes were recently found to be involved in the development of DCM. The mitochondrial calcium uptake 1 gene was shown to alleviate DCM through the mitochondrial Ca2+-dependent antioxidant response.13 Yang et al also suggested that the fibroblast growth factor-21 gene prevents DCM through AMPK (Adenosine 5'-monophosphate (AMP)-activated protein kinase)-mediated antioxidation and lipid-lowering effects in the heart.14 Additionally, rutin, a natural bioflavonoid, was demonstrated to reduce cardiac remodeling and improve myocardial function in a diabetic apolipoprotein E (ApoE) knockout mouse model.15 The programmed cell death 4 gene (PDCD4) has been associated with tumor suppression,16 17 inflammation, and apoptosis in recent years.18–20 It is also closely associated with glycolipid metabolism and inflammation.21 Its absence not only inhibits the development of type 1 diabetes18 and obesity21 by regulating insulin resistance and apoptosis but also regulates lipopolysaccharide-induced inflammation.18 22 Further investigation is required to determine whether PDCD4 plays an important role in type 2 DCM and its specific mechanism.
The PI3K-AKT signal transduction pathway plays a major regulatory role in insulin metabolism.23 A previous study demonstrated that PDCD4 can significantly inhibit PI3K and AKT activation, while PDCD4 overexpression leads to downregulation of phosphorylated PI3K and AKT. Deletion of the PDCD4 gene can significantly upregulate phosphorylation and activity of PI3K and AKT.24 Therefore, we speculated that PDCD4 may block the insulin signaling pathway through the PI3K-AKT signal transduction pathway, thereby aggravating the processes involved in the progression of DCM.
Research design and methods
Ethics statement
All experimental procedures were performed according to the recommendations in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee (approval number: DWLL-2018–019) and Scientific Investigation Board of Shandong University Qilu Hospital (Jinan, China).
Animal model and protocol
Male 12-week-old Sprague-Dawley rats and PDCD4-deficient (PDCD4-/-) rats were obtained from Beijing Viewsolid Biotech (Beijing, China). Then divided them into four groups (n=12 for each group): wild-type non-diabetic (control), PDCD4-/- non-diabetic (control), wild-type diabetic, and PDCD4-/- diabetic. Type 2 diabetes rats was induced by feeding high-fat diet (45% fat and 0.25% cholesterol) for 4 weeks followed by a single intraperitoneal injection of low-dose streptozotocin (STZ; 27.5 mg/kg, Sigma-Aldrich, St. Louis, Missouri, USA) dissolved in citrate buffer (0.1 mol/L, pH 4.5). The control groups were fed normal chow and injected with an identical volume of sodium citrate buffer after 4 weeks. Thereafter, control rats were fed normal chow and diabetic rats were fed the high-fat diet for 20 weeks. The rats were then euthanized and their hearts were excised and fixed in 10% formalin and processed for histological/immunohistochemical analyses, or flash-frozen for molecular biochemical analyses.
The intraperitoneal glucose tolerance test (IPGTT) and insulin tolerance test (IPITT) were performed on all rats by introducing glucose (2 mg/g body weight) or insulin (1 U/kg body weight) by intraperitoneal injection, respectively. The blood glucose level was measured before injection (0 min) and 15, 30, 60, and 120 min after injection. Only rats with blood glucose levels ≥11.1 mM were included in the diabetic groups.
Serum total cholesterol (TC), total triglyceride (TG), free fatty acids (FFA), fasting blood glucose (FBG), and serum fasting insulin (FINS) were measured in all rats using the relevant commercial kits (Roche Diagnostics, Mannheim, Germany).
Echocardiographic examination
After anesthesia with isoflurane, we used the Vevo770 imaging system (Visual Sonics, Toronto, Canada) to perform transthoracic echocardiography on all rats. We employed M-mode echocardiography to measure the left ventricular end-diastolic diameter (LVEDD), left ventricular ejection fraction (LVEF), and fractional shortening (FS) in the LV (LVFS) long axis view. The early (E) and late (A) diastolic mitral flow velocities were measured by pulsed Doppler in the four-chamber view and the ratio of E/A was calculated. Tissue Doppler imaging in the four-chamber view was used to measure the early (E’) and late (A’) diastolic mitral annular velocities and the ratio of E’/A’ was calculated.
Histological and immunohistochemical staining
Excised hearts were fixed in 10% formalin, paraffin-embedded, sectioned into 5-μm-thick sections, placed on slides, and then stained with H&E for cardiomyocyte morphological analysis, and Masson’s trichrome and Sirius Red staining for myocardial fibrosis examination. Sections were incubated with primary antibodies, including interleukin (IL)-6 (1:200, Abcam, Cambridge, UK), tumor necrosis factor-α (1:200, Abcam), IL-10 (1:200, Abcam), collagen I (1:200, Abcam), collagen III (1:200, Abcam) and transforming growth factor beta 1 (TGF)-β1 (1:200, Abcam) overnight at 4°C. Subsequently, horseradish peroxidase-conjugated secondary antibodies were added the next day for 1 hour at 25°C. The stained sections were developed with diaminobenzidine (ZSGB-Bio, Beijing, China) for color development and nuclei were counterstained with hematoxylin. Analysis was conducted using Image-Pro Plus 6.0 software (Media Cybernetics, Rockville, Maryland, USA). Sections were reacted with non-immune IgG secondary antibody only, and no primary and secondary antibodies were used as negative controls.
Quantitative real-time reverse transcriptase PCR (RT-PCR)
Total RNA was extracted from excised heart tissues and H9c2 cardiomyocytes using the RNeasy mini kit (Qiagen, Hilden, Germany). Quantitative real-time RT-PCR was performed using the LightCycler 480 system (Roche Diagnostics) and cDNA Reverse Transcription and SYBR RT-PCR (Takara Bio, Kusatsu, Japan) kits according to the instructions of the manufacturers. cDNA was synthesized using the Prime Script RT reagent kit (Takara Bio) according to the manufacturer’s protocol. Threshold cycle values (Ct) and relative mRNA expression levels were analyzed by the 2-△△Ct method.25 The primers are shown in data.
Western blot analysis
Protein was extracted from excised rat hearts and H9c2 cardiomyocytes using a Total Protein Extraction Kit (Invent Biotechnologies, Plymouth, Minnesota, USA) following the manufacturer’s instructions. Proteins were then separated by sodium dodecyl sulfate and polyacrylamide gel, transferred to polyvinylidene fluoride membranes, and incubated with primary antibodies purchased from Cell Signaling Technologies (Danvers, Massachusetts, USA) at a dilution of 1:1000: PDCD4, PI3K, phosphorylated PI3K, AKT, phosphorylated AKT, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:1000). Membranes were incubated with primary antibodies overnight at 4°C, thereafter, appropriate secondary antibodies (1:5000; Beijing Zhongshan Biotechnology, Beijing, China) were added for 1 hour at room temperature. Transferred blots were visualized using the Immobilon Western HRP Substrate chemiluminescent reagent (Millipore, Darmstadt, Germany). Protein levels were normalized to that of GAPDH.
Cell culture
H9c2 cardiomyocytes (ATCC, Manassas, Virginia, USA), subclones of an original clonal cell line derived from embryonic BD1X rat heart tissue that exhibits many of the properties of skeletal muscle, were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C. Cardiac fibroblasts (CFs) were prepared from rat heart ventricles according to a previously described method.26 27 Cells were collected after digesting the minced LV free wall with trypsin (6 mg/mL) and collagenase type 2 (100 U/mL), and preincubated in DMEM with 10% FBS for 2 hour to separate CFs from myocytes. The cells were then cultured in DMEM supplemented with 10% FBS in a humidified incubator with 5% CO2 and 95% air at 37°C.
Adenovirus-mediated overexpression and siRNA transfection
For the overexpression experiments, adenovirus carrying recombination PDCD4 (rPDCD4) and green fluorescent protein (GFP) (pAdxsi-GFP-rPDCD4; SinoGenoMax, Beijing, China) was diluted in DMEM and added to the cell culture media (DMEM containing 10% FBS) according to the multiplicity of infection. For the knockdown experiments, Shanghai GenePharma (Shanghai, China) was commissioned to design four pairs of small interfer RNA (siRNA) targeting the rat PDCD4 gene, and the sequence with the highest silencing efficiency was selected after screening (No 553: 5′-GCGGAGATGTTAAGGGATTTG dtdt-3′). Adenovirus carrying rPDCD4-short hairpin RNA (shRNA) and GFP (pAdxsi-GFP-rPDCD4-553 shRNA) was used to transfect H9c2 cardiac myocytes and myocardial fibroblasts. The amount of culture medium mixed with virus required for adenovirus infection was 1 mL/well for the 6-well plate, 0.5 mL/well for the 12-well plate, and 0.2 mL/well for the 24-well plate.
TdT-mediated dUTP nick end labeling (TUNEL) assay
We obtained myocardial samples from the four rat groups to determine the extent of apoptosis using an In Situ Cell Death Detection Kit, TMR red (Roche), according to the manufacturer’s instructions. Nuclei were stained with 4',6-diamidino-2-phenylindole. The apoptosis ratio was calculated as the proportion of apoptotic cells to the total number of cells in the myocardium.
Statistical analysis
Data are presented as mean±SEM or median (IQR). Continuous data were compared using an unpaired Student’s t-test or one-way analysis of variance (ANOVA), after testing for normality and equality of variance. Significant interactions were observed in ANOVAs and post hoc Bonferroni tests were performed. IBM SPSS Statistics for Windows, V.21.0 (IBM, Armonk, New York, USA) was used to determine the statistical significance of the tests. Values of p<0.05 were considered statistically significant.
Results
PDCD4 was involved in the development of DCM and its deficiency improves abnormal lipid metabolism and insulin resistance in type 2 diabetic rats
Healthy male rats were fed a high-fat diet combined with low-dose STZ to induce an animal model bearing similarity to human type 2 diabetes. We examined PDCD4 mRNA and protein expression in the myocardium of diabetic and control rats. Compared with control hearts, diabetic heart tissues showed a marked increase in PDCD4 mRNA and protein expression (figure 1A,B,E). Meanwhile, PDCD4 expression increased accordingly in H9c2 rat myocardial cells under time-dependent stimulation with high glucose (figure 1C,D,F). We predicted that PDCD4 might be involved in the occurrence and development of DCM.
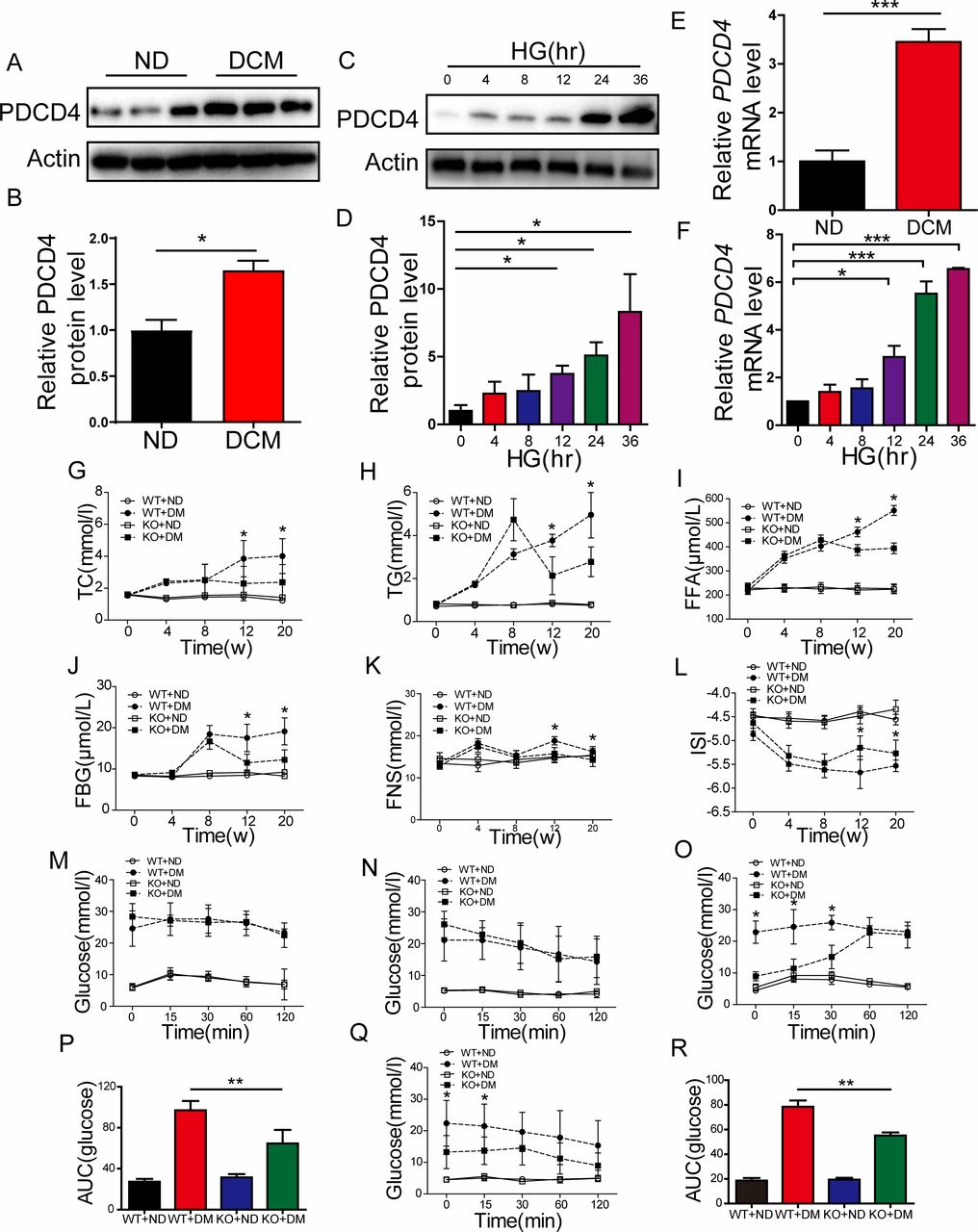
Programmed cell death 4 (PDCD4) was involved in the development of diabetic cardiomyopathy (DCM) and its deficiency improves abnormal lipid metabolism and insulin resistance in type 2 diabetic rats. (A,B) Representative images and quantitative analysis of PDCD4 protein expression in the myocardium of control and diabetic rats. (C–D) Representative images and quantitative analysis of PDCD4 protein expression changes in H9c2 cells accompanied by time-dependent high-glucose (HG) stimulation. (E–F) Quantitative analysis of PDCD4 mRNA expression in the myocardium of control and diabetic rats. (G–K) Quantitative analysis of serum total cholesterol (TC) levels, serum triglyceride (TG) levels, serum free fatty acid (FFA) levels, serum fasting blood glucose (FBG) levels, and serum fasting insulin (FNS) levels in four groups of rats. (L) Quantitative analysis of serum insulin sensitive index (ISI) levels in four groups of rats. (M–N) Quantitative analysis of intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) between wild-type (WT) and PDCD4-/- rats after ingesting a high-fat diet for 8 weeks. (O–P) Quantitative analysis of IPGTT and the area under curve (AUC) between four groups of rats after ingesting a high-fat diet for 12 weeks. (Q–R) Quantitative analysis of IPITT and AUC between four groups of rats after ingesting a high-fat diet for 12 weeks. Data were expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001. DM, diabetes mellitus; KO, knockout; ND, normal diet.
We thus constructed a PDCD4 knockout (PDCD4-/-) rat. Type 2 diabetes was induced as previously described in PCDC4-/- and wild-type rats. The weights of the wild type and PDCD4-/- diabetic rats were both significantly lower (p<0.05) than the control rats after 12 weeks of a high-fat diet, which indicated a successful model. However, no significant difference was observed in the body weights of PDCD4-/- rats compared with wild-type rats in the non-diabetic group, although there appeared to be a decreasing trend (data not shown). After diabetes induction, serum TC, TG, FFA, FBG, and FINS levels were significantly decreased (p<0.05) in PDCD4-/- and wild-type rats after 12 and 20 weeks ingesting a high-fat diet (figure 1G–K). Further, the serum insulin sensitive index (ISI) was significantly increased (figure 1L). Thus, PDCD4 deficiency improved abnormal lipid metabolism in type 2 diabetic rats.
To examine the impact of PDCD4 on insulin resistance of type 2 DCM rats, IPGTTs and IPITTs were conducted, and the area under curve (AUC) for blood glucose was calculated. The IPGTT and IPITT results did not differ for the wild-type and PDCD4-/- diabetic groups at 0 min, 15 min, 30 min, 60 min, and 120 min after 8 weeks ingesting a high-fat diet (figure 1M,N). However, after 12 weeks, the IPGTT result was significantly reduced in the PDCD4-/- diabetic group at 0 min, 15 min, and 30 min (figure 1O), and the AUC was significantly decreased (figure 1P). Meanwhile, the blood glucose value and AUC of the IPITT were significantly reduced at 0 min and 15 min (figure 1Q,R). Thus, PDCD4 deficiency can improve insulin resistance in type 2 diabetic rats.
PDCD4 deficiency can improve LV remodeling and function in type 2 diabetic rats and affects the morphology of cardiac myocytes
B-type natriuretic peptide (BNP), as a quantitative marker of heart failure, reflects both LV systolic and diastolic dysfunction, as well as valvular dysfunction. In contrast to wild-type diabetic rats, BNP mRNA decreased significantly in the PDCD4-/- diabetic group (figure 2A). After successful diabetic modeling, echocardiographic measurements (figure 2B) showed that, compared with the control group, the LVEF, LVFS, E/A ratio, and E'/A' ratio decreased significantly and LVEDD, LVESD, and E/E' ratio increased significantly in the diabetic group (figure 2C–I). In comparison with the wild-type diabetic group, LVEF, LVFS, E/A ratio, and E’/A’ ratio were significantly increased and LVEDD, LVESD, and E/E' ratio decreased in the PDCD4-/- diabetic group (figure 2C–I). These results suggest that PDCD4 gene knockout can improve LV function in DCM rats.

Programmed cell death 4 (PDCD4) deficiency can improve left ventricular remodeling and function in type 2 diabetic rats and affects the morphological changes of cardiac myocytes. (A) Quantitative analysis of B-type natriuretic peptide (BNP) mRNA expression between four groups of rats. (B) Representative echocardiographic images of four groups of rats. (B-1) Two-dimensional echocardiogram showing left ventricular long-axis view; (B-2) M-mode echocardiogram showing left ventricular dimensions; (B-3) Pulse-wave Doppler echocardiogram depicting diastolic mitral flow; (B-4) Tissue Doppler echocardiogram displaying mitral annular velocities. (C) Quantitative analysis of left ventricular ejection fraction (LVEF) in four groups of rats. (D) Quantitative analysis of left ventricular fractional shortening (LVFS) in four groups of rats. (E) Quantitative analysis of left ventricular end-diastolic diameter (LVEDD) in four groups of rats. (F) Quantitative analysis of left ventricular end-systolic diameter (LVESD) in four groups of rats. (G) Quantitative analysis of the ratio of early to late diastolic mitral flow velocities (E/A) in four groups of rats. (H) Quantitative analysis of the ratio of early to late diastolic mitral annular velocities (E′/A′) in four groups of rats. (I) Quantitative analysis of the ratio of early diastolic mitral flow to early diastolic mitral annular velocities (E/E′) in four groups of rats. (J) Representative H&E staining of myocardial cross sections in four groups of rats (scale bar: 100 µm). (K) Quantitative analysis of the surface area changes of H9c2 cardiac myocytes in six groups of cells through immunohistochemical staining of myosin heavy chain measurement. Data were expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001. DM, diabetes mellitus; HG,high glucose; KO, knockout; MOCK, mock-vehicle transfection group; ND, normal diet; NG, normal glucose; OC, osmoticcontrol; SiP, SiRNA-PDCD4; siRNA, small interfer RNA; WT, wild type.
Meanwhile, we applied H&E staining to sections of myocardium from the left ventricles (figure 2J). We observed dense, neatly arranged myocardium cells with a clear structure, uniform nucleus size, and even cytoplasm staining in the control group fed a normal diet. However, the myocardial cells in the wild-type diabetic group were hypertrophic, distorted, and disordered in arrangement, with irregular nuclear size and many myocardial fiber fractures, and the gap junctions were significantly enlarged. Comparatively, the myocardial cells in the PDCD4-/- diabetic group were smaller and less distorted and fractured, exhibiting a relatively neat and compact arrangement with a clear structure, and the intercellular space was reduced (figure 2J). In vitro, high-glucose stimulation significantly increased the surface area of cardiac myocytes, while normalized by silencing PDCD4 expression through siRNA virus transfection. Compared with the PDCD4 siRNA virus transfection, cardiomyocyte surface area increased significantly after PDCD4 overexpressed adenovirus transfection, as observed by measurement of myosin heavy chains through immunohistochemical staining (figure 2K).
PDCD4 deficiency attenuates myocardial fibrosis and inflammation in diabetic rats
Masson’s trichrome and Sirius Red staining demonstrated that the PDCD4-/- diabetic group displayed a reduced fibrotic area (collagen deposition) in the myocardial matrix and perivascular region compared with the wild-type diabetic group; the arrangement was more orderly with reduced perivascular fibrosis (figure 3A,B,I1–I2). Immunohistochemical analysis revealed more intensive staining of collagen I, collagen III, and TGF-β1 in wild-type diabetic rat hearts than was observed in the PDCD4-/- diabetic rat hearts (figure 3C–E,I3–I5). RT-PCR analysis also revealed greater mRNA expression of collagen I, collagen III, and TGF-β1 in wild-type diabetic rat hearts than in the PDCD4-/- diabetic rat hearts (figure 3J).
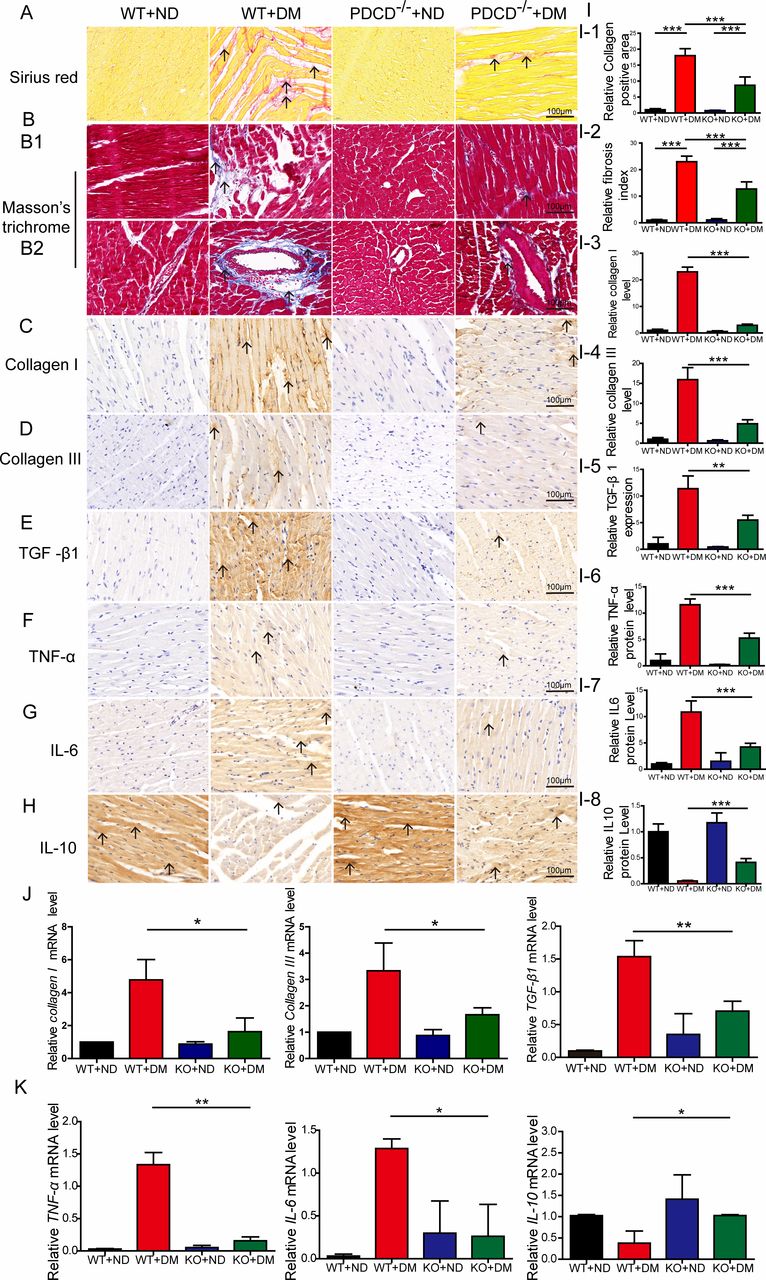
Programmed cell death 4 (PDCD4) deficiency attenuates myocardial fibrosis and inflammation in diabetic rats. (A, I-1) Representative images and quantitative analysis of Sirius Red staining of myocardial fibers (scale bar: 100 µm). (B, I-2) Representative images and quantitative analysis of Masson’s trichrome staining of myocardial fibers (scale bar: 100 µm). (B1) Representative images of Masson’s trichrome staining of myocardial matrix fibers (scale bar: 100 µm). (B2) Representative images of Masson’s trichrome staining of perivascular fibers (scale bar: 100 µm). (C–E, I-3–I-5) Representative immunohistochemical staining and quantitative analysis of Collagen I, Collagen III, and transforming growth factor (TGF)-β1 protein expression in the myocardium of four groups of rats (scale bar: 100 µm). (J) Quantitative analysis of Collagen I, Collagen III, and TGF-β1 mRNA expression in the myocardium of four groups of rats. (F–H, I-6–I-8) Representative immunohistochemical staining and quantitative analysis of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-10 protein expression in the myocardium of four groups of rats (scale bar: 100 µm). (K) Quantitative analysis of TNF-α, IL-6, and IL-10 mRNA expression in the myocardium of four groups of rats. Data were expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001. DM, diabetes mellitus; KO, knockout; ND, normal diet; WT, wild type.
Immunohistochemical analysis indicated that proinflammatory factors TNF-α and IL-6 were significantly reduced, while anti-inflammatory cytokine expression (IL-10) increased (figure 3F–H,I6–I8) in the PDCD4-/- diabetic group, compared with the wild-type diabetic group. Meanwhile, RT-PCR analysis revealed the same changes in the PDCD4-/- diabetic group compared with the wild-type diabetic (figure 3K). These results indicate that PDCD4 deficiency improves myocardial inflammation in diabetic rats.
PDCD4 expression intervention affects proliferation, transformation, and oxidative stress of CFs stimulated by high glucose in vitro
We examined the ratio of Ki67-positive cells to reflect fibroblast proliferation in vitro. The proportion of alpha-smooth muscle actin (α-SMA)-positive cells in S100A4-positive cells reflects the transformation from CFs to myofibroblasts. The results indicated that high-glucose stimulation substantially promoted fibroblast proliferation, as shown by the increased proportion of Ki67-positive cells. The silencing of PDCD4 expression through siRNA virus transfection may inhibit fibroblast proliferation. Conversely, fibroblast proliferation was significantly enhanced after transfection of PDCD4 overexpressed adenovirus, compared with that of the PDCD4 siRNA virus (figure 4A,C-1).
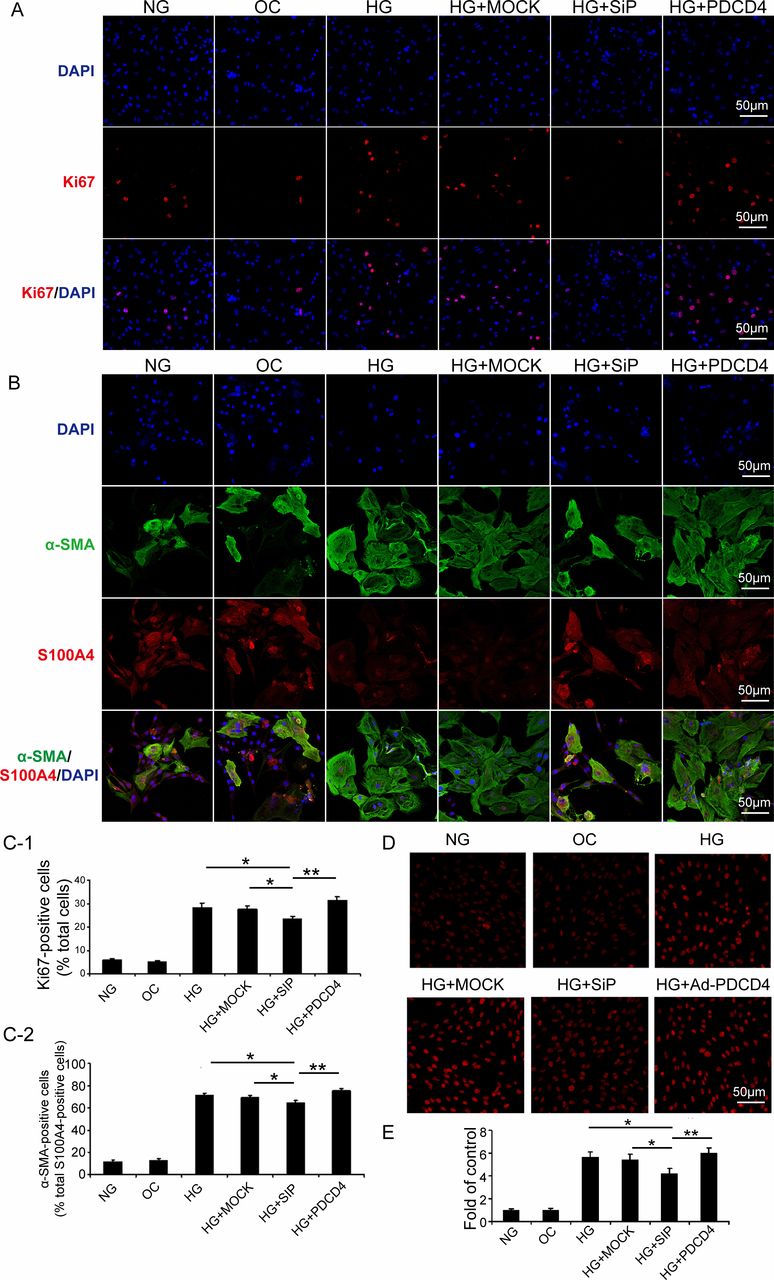
Programmed cell death 4 (PDCD4) expression intervention affects proliferation, transformation, and oxidative stress of cardiac fibroblasts stimulated by HG in vitro. (A) Immunofluorescence analysis of six groups of cells expressing Ki67 (red) and nuclei stained with DAPI (blue) (scale bar: 50 µm). (C-1) Quantification of Ki67-positive cells. (B) Immunofluorescence analysis of six groups of cells expressing S100A4 (red), α-SMA (blue), and nuclei stained with DAPI (blue) (scale bar: 50 µm). (C-2) Quantification of α-SMA and S100A4, both positive cells. (D) Representative dihydroethidium (DHE) staining of apoptosis in six groups of cells (scale bar: 50 µm). (E) Quantification of DHE-positive cells in six groups of cells. Data were expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001. DAPI, 4',6-diamidino-2-phenylindole; HG, high glucose; MOCK, mock-vehicle transfection group; NG, normal glucose; OC, osmotic control; SiP, SiRNA-PDCD4; siRNA, small interfer RNA; α-SMA, alpha-smooth muscle actin.
High-glucose stimulation promoted the transformation of CFs to myofibroblasts, which was manifested by the increased proportion of α-SMA-positive cells in S100A4-positive cells. PDCD4 silencing, by siRNA virus transfection, could inhibit CF transformation. However, PDCD4 overexpressed adenovirus transfection significantly enhanced transformation, compared with that of the PDCD4 siRNA virus transfection (figure 4B,C-2).
After high-glucose stimulation, PDCD4 overexpression, and silencing, we adopted dihydroethidium staining to examine the oxidative stress of rat myocardial fibroblasts (O2 content alterations). The results revealed that high-glucose stimulation upregulated O2 content in the cell, while siRNA virus transfection (PDCD4 silencing) reduced O2 content, and PDCD4 overexpression recovered O2 content with a significant increase (figure 4D,E).
PDCD4 regulated myocardial apoptosis and the PI3K-AKT pathway in vivo and in vitro
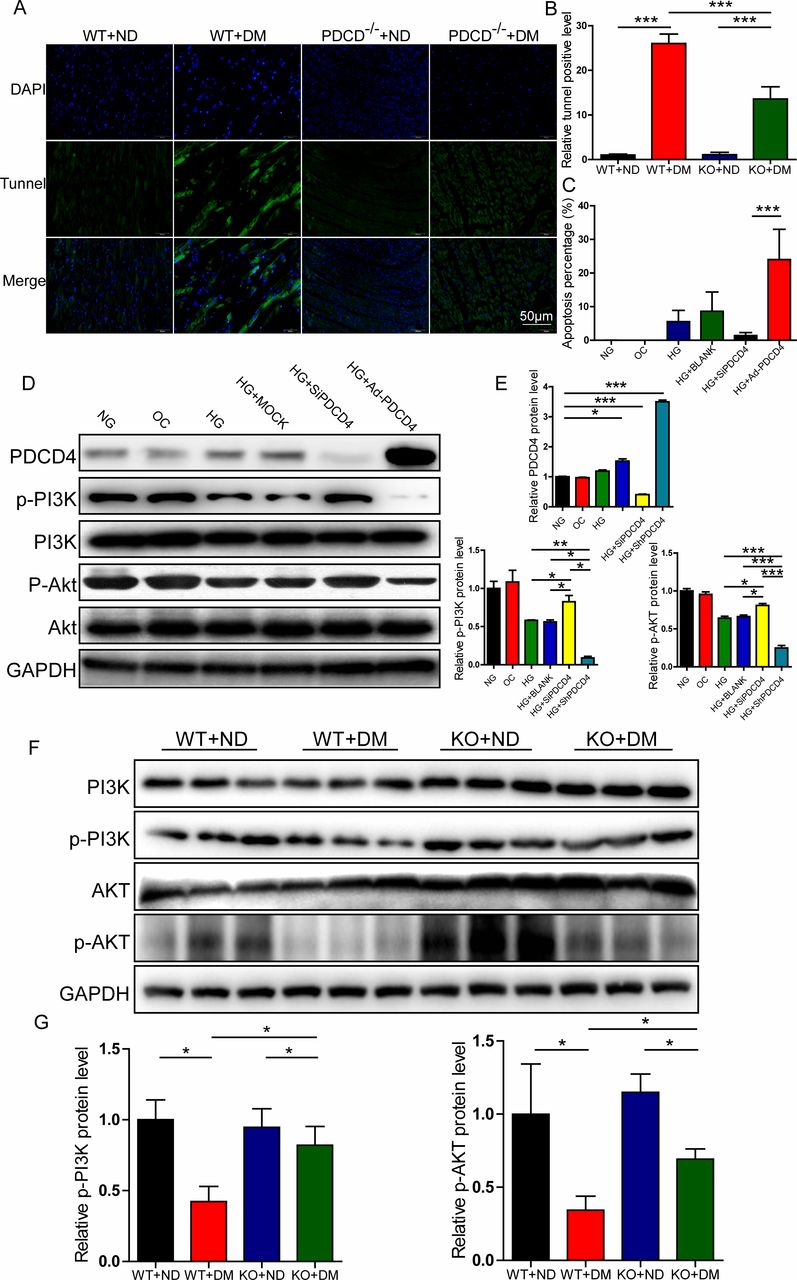
After 20 weeks ingesting a high-fat diet, the apoptotic rate of LV myocardial cells was measured by TUNEL methods. The results indicated that the number of apoptotic cells in PDCD4-/- diabetic rats was significantly reduced compared with wild-type diabetic rats (figure 5A,B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Programmed cell death 4 (PDCD4) regulated myocardial apoptosis and PI3K-AKT pathway in vivo and in vitro. (A) Representative TUNEL staining of apoptosis in the myocardium of four groups of rats (scale bar: 50 µm). (B) Quantification analysis of TUNEL-positive cells in four groups of rats. (C) Quantification analysis of flow cytometry results of apoptosis in six groups of cells. (D) Representative western blot of PDCD4, pPI3K, pAKT, PI3K, and AKT expression in six groups of cells. (E) Quantification analysis of PDCD4, pPI3K, and pAKT protein expression in six groups of cells. (F) Representative western blot of pPI3K, PI3K, pAKT, and AKT expression in the myocardium of four groups of rats. (G) Quantification analysis of PDCD4, pPI3K, and pAKT protein expression in the myocardium of four groups of rats. Data were expressed as mean±SEM. *p<0.05, **p<0.01, ***p<0.001. AKT, protein kinase B; DAPI, 4',6-diamidino-2-phenylindole; DM, diabetes mellitus; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HG,high glucose; KO, knockout; MOCK, mock-vehicle transfection group; ND, normal diet; NG, normal glucose; OC, osmoticcontrol; PI3K, phosphatidylinositol 3-kinase; SiP, SiRNA-PDCD4; SiRNA, small interfer RNA; WT, wild type.
High-glucose stimulation significantly improved myocardial H9c2 cell apoptosis, and the effect was inhibited by transfection with PDCD4 siRNA viruses (figure 5C).
Based on the above results, we investigated ratios of p-PI3K/PI3K and p-AKT/AKT, which are the major insulin resistance pathway molecules. In vitro, p-PI3K and p-AKT levels were significantly reduced after stimulation with high glucose, but rescued after PDCD4 silencing. Additionally, p-PI3K and p-AKT levels were significantly lower than those observed in the PDCD4 overexpressed adenovirus transfection group (figure 5D,E). In vivo, compared with the wild-type diabetic group, the ratios of p-PI3K and p-AKT in the PDCD4-/- diabetic group were significantly enhanced, indicating that PDCD4 gene knockout improved the insulin resistance pathway and upregulated insulin sensitivity (figure 5F,G).
Discussion
Epidemiological data show that diabetes has become a global epidemic, and DCM as a major cause of heart failure and death is gaining increasing attention.1 9 11 PDCD4 has recently been recognized as a novel inflammation and apoptosis gene18 and is closely associated with type 2 diabetes.21 However, the role PDCD4 plays in type 2 DCM, and the associated mechanism, is unclear. We established a type 2 diabetes rat model and observed increased levels of PDCD4 in myocardial tissue from the type 2 DCM rats hearts compared with controls. Meanwhile, the PDCD4 expression level was significantly upregulated with high-glucose stimulation in the rat myocardial cell line H9c2 in vitro. Therefore, we speculated that PDCD4 may be involved in the occurrence and development of DCM.
Thereafter, we constructed PDCD4 gene knockout rats. Wild-type and PDCD4 knockout rats were selected to establish the type 2 diabetes model28 concurrently. Body weights of wild-type and PDCD4-/- diabetic rats did not differ significantly. Meanwhile, compared with the wild-type diabetic group, the TC, TG, FFA, FBG, and FINS results of the PDCD4-/- diabetic group were significantly reduced, and serum ISI was significantly increased after ingesting a high-fat diet for 12 and 20 weeks. The IPGTT and IPITT blood glucose values and AUC from the PDCD4-/- diabetic group were both significantly reduced. Thus, it was suggested that the lack of PDCD4 gene expression can significantly improve abnormal lipid metabolism and insulin resistance, which can induce cardiovascular damage.7
Type 2 diabetes mellitus can alter cardiac structure and function,4 and LV remodeling and dysfunction are typical features of DCM.11 Echocardiography detected significantly increased LVEF, FS, E/A and E'/A' values for the PDCD4-/- diabetic group compared with the wild-type diabetic group. As one of the indicators of cardiac function, BNP also decreased significantly in the PDCD4-/- diabetic group. These results suggest that PDCD4 gene knockout can improve myocardial function in type 2 DCM rats. Meanwhile, H&E staining of a section of myocardium from the LV indicated that myocardial cells in the control group were dense, and arranged neatly, with clear structure, uniform nucleus size, and even cytoplasm staining. Interestingly, we observed decreased myocardial cell size in the PDCD4-/- diabetic group, and the myocardial cells were less distorted and fractured with a relatively neat and compact arrangement, relatively clear structure, and reduced intercellular space compared with the wild-type diabetic group. In vitro, high-glucose stimulation significantly increased the surface area of cardiac myocytes, while silencing of PDCD4 expression, caused by siRNA virus transfection, normalized the surface area. Meanwhile, compared with the PDCD4 siRNA virus transfection group, cardiac myocyte surface area increased significantly post-PDCD4 overexpressed adenovirus transfection.
Previous research has shown that altered myocardial fibrosis,9 increased inflammation, higher induction of apoptosis, and increased oxidative stress2 10 represent the underlying mechanisms involved in the development of type 2 DCM.5 11 PDCD4-deficient rats are known to be resistant to inflammation in STZ-induced type 2 diabetes, but the relationship was unclear in DCM. Through Masson’s trichrome, Sirius Red staining, fibrosis-related collagen I, III immunohistochemical staining, and RT-PCR, we demonstrated that collagen deposition, fibrogenic factors, and TGF-β129 levels were significantly reduced after PDCD4 knockout in vivo, with relatively regular cellular arrangement and reduced perivascular fibrosis. Inflammation is a primary characteristic in diabetes.12 30 We discovered significantly decreased expression of proinflammatory factors after PDCD4 knockout in vivo, while expression of IL-10, an anti-inflammatory factor, was significantly increased. Meanwhile, TUNEL analysis was performed to determine the rate of myocyte apoptosis, which participates in the development of DCM5 and found that compared with control rats, PDCD4-/- rats exhibited significantly reduced numbers of apoptotic cells. Flow cytometry was used to detect myocardial cell apoptosis in H9c2 cells in vitro. High-glucose stimulation was shown to significantly enhance myocardial cell apoptosis, and this effect was altered by PDCD4 silencing. Diabetic myocardial tissue of PDCD4 knockout rats displayed significantly reduced apoptosis and oxidative stress. Therefore, it can be concluded that PDCD4 is involved in the occurrence and development of DCM fibrosis and inflammation, and this may contribute to the improvement of LV remodeling and deficiency.
Cardiac tissue remodeling is regulated by multiple cell types, including cardiomyocytes, fibroblast endothelial cells, smooth muscle cells, and hematopoietic-derived cells.29 As the main effector cells of cardiac fibrosis, CFs are an important cellular component of the myocardial response to injury and hypertrophic stimuli, which regulate the synthesis and degradation of the extracellular matrix.31 32 Silencing of PDCD4 (via transfection with siRNA viruses) can decrease the O2 content, and overexpression of PDCD4 (via transfection with PDCD4-overexpressed adenovirus) can induce a significant O2 increase in rat primary CFs. Thus, PDCD4 intervention can improve the oxidative stress conditions in myocardial fibroblasts. The critical event in the evolution of cardiac fibrosis is fibroblast proliferation and conversion from myocardial fibroblasts to myofibroblasts with increased percentages of Ki67 and α-SMA-positive cells in S100A4-positive cells.33–36 PDCD4 silencing can inhibit fibroblast proliferation, while PDCD4 overexpression can enhance fibroblast proliferation. Therefore, we can conclude that PCDC4 silencing can significantly inhibit the proliferation and transdifferentiation of rat myocardial fibroblasts, thus inhibiting the development of DCM.
Based on the above data, we further investigated PDCD4 signaling pathways involved in insulin resistance of DCM in vivo and in vitro. Previous studies have reported that the PI3K/AKT11 signaling pathway is closely associated with diabetic cardiac dysfunction and responsible for most of the metabolic actions of insulin.7 Microvascular dysfunction may also play an important role for whole heart measurement of PI3K and AKT phosphorylation in the context of fibrosis and inflammation. The contractile kinetics should be assayed if the dysfunction is related to cardiomycyte insulin sensitivity. PDCD4 knockout can significantly improve inhibition of the PI3K/AKT signaling pathway induced by insulin resistance, increase AKT phosphorylation, and thus upregulate insulin sensitivity. This would significantly improve abnormal glucose and lipid tolerance, and insulin resistance seen in type 2 diabetic rats.
Limitations and future directions
There were several potential limitations in the current study. First, we did not explore the role of other signaling pathways including miRNAs in insulin resistance, apart from the PI3K/AKT pathway, to mediate the beneficial effect of PDCD4 inhibition on DCM. For example, the PDCD4 protein synthesis could be inhibited by microRNAs including miR21.37 Second, it remains to be investigated whether insulin resistance drugs such as metformin could inhibit or reduce PDCD4 expression or protect beta cells to improve DCM, as well as the effects of possible drug molecules. Third, further experiments are warranted to clarify the exact molecular mechanism of PDCD4 gene knockout for alleviating cardiac remodeling and myocardial function.
Conclusion
In conclusion, PDCD4 expression intervention can significantly improve the PI3K-AKT signaling pathways relative to insulin resistance, leading to alleviated lipid metabolism disorders, and decreased inflammation and interstitial fibrosis. This would eventually improve type 2 DCM systolic and diastolic dysfunction, preventing or delaying the development of DCM and heart failure. The PDCD4 mechanism involved inflammation, remodeling, and apoptosis of CFs and myocardial cells. These results provide a new therapeutic avenue, as well as novel research ideas, for improving the prognosis of patients with type 2 DCM. Development of a therapeutic inhibitor for PDCD4 gene expression could pathophysiologically and pharmacologically prevent type 2 DCM. Further studies need to confirm our preliminary findings.
Acknowledgments
We thank Dr Changhong Sun (Beijing Viewsolid Biotech, China) for providing the construction of knockout rats and Shangwen Sun (Shandong University, China) for echocardiographic analysis in this study.
References
Footnotes
Contributors JZ contributed to data research, analysis, and manuscript writing. ZY was responsible for rat breeding. SH, XWu, NL, LC, XWa, and QL contributed to data collection and research. MZ reviewed and edited the manuscript. FG contributed to the study concept and revision and final approval of the manuscript.
Funding The work was supported by research grants from the National Natural Science Foundation of China (81701404 and 81470403), the Key Research and Development Plan and Natural Science Foundation of Shandong Province (ZR2017BH074, ZR2016HM01, ZR201910290090), and the China Postdoctoral Science Foundation (2017M610431).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval This study was approved by the Ethics Committee (approval no DWLL-2018-019) and Scientific Investigation Board of Shandong University Qilu Hospital (Jinan, China).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are available upon reasonable request.