Article Text

Abstract
Introduction Diabetic retinopathy is a major complication of diabetes recently associated with compromised photoreceptor function. Multiple stressors in diabetes, such as hyperglycemia, oxidative stress and inflammatory factors, have been identified, but systemic effects of diabetes on outer retina function are incompletely understood. We assessed photoreceptor physiology in vivo and in isolated retinas to better understand how alterations in the cellular environment compared with intrinsic cellular/molecular properties of the photoreceptors, affect light signal transduction and transmission in the retina in chronic type 2 diabetes.
Research design and methods Photoreceptor function was assessed in BKS.Cs-Dock7m+/+Lepr db/J mice, using homozygotes for Leprdb as a model of type 2 diabetes and heterozygotes as non-diabetic controls. In vivo electroretinogram (ERG) was recorded in dark-adapted mice at both 3 and 6 months of age. For ex vivo ERG, isolated retinas were superfused with oxygenated Ames’ media supplemented with 30 mM glucose or mannitol as iso-osmotic control and electrical responses to light stimuli were recorded.
Results We found that both transduction and transmission of light signals by rod photoreceptors were compromised in 6-month-old (n=9–10 eyes from 5 animals, ***p<0.001) but not in 3-month-old diabetic mice in vivo (n=4–8 eyes from 2 to 4 animals). In contrast, rod signaling was similar in isolated retinas from 6-month-old control and diabetic mice under normoglycemic conditions (n=11). Acutely elevated glucose ex vivo increased light-evoked rod photoreceptor responses in control mice (n=11, ***p<0.001), but did not affect light responses in diabetic mice (n=11).
Conclusions Our data suggest that long-term diabetes does not irreversibly change the ability of rod photoreceptors to transduce and mediate light signals. However, type 2 diabetes appears to induce adaptational changes in the rods that render them less sensitive to increased availability of glucose.
- diabetic retinopathy
- electrophysiology
- glucose
- retina
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Retinal neuronal dysfunction in diabetic retinopathy frequently precedes microvascular changes. Damage to photoreceptors has been reported, although the literature is not entirely conclusive, and the underlying mechanisms and reversibility of photoreceptor dysfunction in diabetes remain unknown.
What are the new findings?
We confirm previous studies showing that photoreceptor and ON-bipolar cell amplitudes are reduced and implicit times are delayed in vivo in diabetic mice in an age-dependent manner. Conversely, we report normal amplitudes and implicit times of the isolated diabetic retina under optimal ex vivo conditions. Elevated glucose concentrations augment photoreceptor function in the isolated 6-month-old non-diabetic retina, but this response is impaired in diabetic retinas.
How might these results change the focus of research or clinical practice?
Our results clarify that photoreceptor function is not irreversibly damaged and that systemic factors may suppress photoreceptor function in diabetic mice in vivo. The consequences of altered glucose handling in long-term diabetic photoreceptors remain to be evaluated.
Introduction
Diabetic retinopathy, one of the leading causes of irreversible vision loss worldwide,1 has traditionally been characterized by retinal microvascular abnormalities. Recent studies in human patients with diabetes suggest, however, that retinal diabetic neuropathy with neuroglial dysfunction and degeneration frequently precedes visible microvasculopathy.2 3 Even in the absence of, or with minimal signs of retinal vascular abnormalities, patients with diabetes show dysfunction and loss of retinal neuronal cells. In fact, impairment of the neurovascular unit has been suggested to lead to or accelerate retinal vascular disease progression since implicit time delay of the multifocal electroretinogram (ERG) predicts onset of visible vascular changes.4 Damage to nerve fiber, ganglion cell and inner plexiform layers in the human eye has been demonstrated in diabetes by impaired pattern ERG and oscillatory potentials5 and thinning of inner retinal layers.3 Reduced amplitudes and longer implicit times of the scotopic and photopic b-waves5 also implicate dysfunction of rod and cone bipolar cells in retinal diabetic neuropathy of human patients. These findings are not universal, however, and reduced scotopic a-wave and b-wave amplitudes have not been shown in all studies.6
Murine models of diabetes share many features of retinal dysfunction and cell loss with patients with diabetes. The positive component of the scotopic threshold response, a measure of retinal ganglion cell (RGC) function, is attenuated following streptozotocin (STZ)-induced type 1 diabetes in the rat.7 In addition, thickness of the nerve fiber and RGC layer, and RGC density are all reduced in STZ-induced rodents.3 Loss or reduced inhibitory output of amacrine cells has been observed in type 1 diabetes models,8–12 and may underlie reduced amplitudes and delayed implicit times of oscillatory potentials.7 13 Similarly to human studies, outer retinal dysfunction and cell loss in diabetic animal models remain controversial. Rod photoreceptor and bipolar cell dysfunction, based on reduced amplitudes and delayed implicit times of the scotopic a-waves and b-waves, were reported in the db/db mouse, a type 2 diabetes model.14 Photoreceptor death was also reported in a rat STZ model,15 but was not validated in other studies16 17 which reported no or only minor changes in the scotopic ERG,7 or even increased photoreceptor function in Zucker Diabetic Fatty (ZDF) rats, another type 2 diabetes model.18
Several studies have implicated photoreceptors in the pathogenesis of diabetic retinopathy.19 Rod photoreceptors are a significant source of oxidative stress in diabetic animals17 and rod degeneration or blocking of visual transduction protects from vascular diabetic retinopathy.20–24 However, both systemic and cell-intrinsic mechanisms affecting photoreceptor function during diabetes are not well understood. Transient hyperglycemia in patients with diabetes has been associated with increased scotopic b-wave and photopic a-wave and b-wave amplitudes.25 While the multifocal ERG waveform is delayed in normoglycemic patients with diabetes compared with healthy volunteers, this is partly reversed by elevation of blood glucose to their usual level of (habitual) hyperglycemia.26 27 These findings indicate that retinal signaling is modulated by both acute systemic hyperglycemia and photoreceptor-intrinsic changes by chronic diabetes. We provide evidence for our hypothesis that under ideal recording conditions long-term uncontrolled type 2 diabetes in db/db mice does not permanently disrupt cell-intrinsic mechanisms in rod photoreceptors that are responsible for transducing and transmitting light signals. This suggests that attenuation of the in vivo ERG in diabetic mice is likely due to diabetes-induced systemic factors influencing rod photoreceptors, which once removed, can no longer mediate suppression of photoreceptor responses in the context of the ex vivo testing. However, we find that rod photoreceptors in the ex vivo isolated diabetic retina are less sensitive to acute elevation of extracellular glucose, likely due to changes at the cellular level.
Research design and methods
Animals
Male and female C57B/6J mice and BKS.Cg-Dock7m+/+Leprdb/J mice were bred in house or purchased at 8–10 weeks of age from Jackson Laboratory. Animals homozygous for the Leprdb mutation (db/db) are diabetic. Age-matched heterozygotes (db/+) were used as non-diabetic controls. All experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Weight, blood glucose and HbA1c
Animals were kept under a 12:12 hour light/dark cycle and given ad libitum access to food and water. Diabetic status was confirmed by measuring weight, blood glucose and HbA1c at 3 or 6 months of age (online supplementary figure 1).
Supplemental material
Blood glucose concentrations during isoflurane anesthesia were assessed in C57B/6J mice immediately before anesthesia and 20 min after beginning inhalation of 2% isoflurane.
In vivo ERG
Mice were fully dark adapted and anesthetized with 1.8%–3.0% isoflurane in room air using a SomnoSuite Low Flow Anesthesia System (Kent Scientific). Pupils were dilated with 2% atropine sulfate eye drops (Akorn, Illinois) and corneas lubricated with a mixture of phosphate buffered saline (PBS) with Goniovisc (2.5% hypromellose solution; Contacare, Gujarat, India). Body temperature was maintained by placement on a heated table throughout the experiment. ERG waveforms were recorded using corneal contact lens electrodes and subdermal needle electrodes between the eyes as reference and at the base of the tail as ground electrodes. Responses to light flashes (rod stimuli, to activate rod phototransduction in rodents) were collected using a ColorDome and Espion V.6 software (Diagnosys LLC).
For scotopic ERGs, 20 sweeps with inter-sweep intervals of 2 s were recorded at light flash energies (luminous energy, QV) from 10−5 to 10−3 cd.s.m−2, 10 sweeps with 5 s intervals at 10−2 cd.s.m−2, 5 sweeps with 10 s intervals from 10−1 to 0.5 cd.s.m−2, 3 sweeps with 20 s intervals from 1 to 5 cd.s.m−2, and 2 sweeps with a 60 s interval at 10 and 30 cd.s.m−2. Oscillatory potentials were high-pass filtered above 25 Hz and subtracted from ERG waveforms. a-wave and b-wave response amplitudes were measured between baseline and trough and between trough and peak respectively, and were plotted as the response amplitude over the log light intensity. a-wave and b-wave implicit times were determined as time from stimulus onset to peak. The amplification constant A of phototransduction at 1 cd.s.m−2 was calculated as described by Lamb and Pugh.28
Ex vivo ERG
Mice were fully dark adapted overnight and sacrificed by CO2 asphyxia. Eyes were enucleated and dissected in Ames’ media supplemented with 0.25 mM sodium glutamate29 and oxygenated with 95% O2/5% CO2. The anterior eye and the lens were removed and retinas were carefully dissected and mounted on an ex vivo specimen holder, as previously described.30 Tissue was superfused with oxygenated Ames’ media containing 0.25 mM sodium glutamate heated to 36°C. Dissected eye cups from fellow eyes were stored in oxygenated Ames’ media at room temperature for up to 2 hours. ERG waveforms were elicited by light stimuli of increasing intensity at 530 nm, amplified using a DP-311 differential amplifier (Warner Systems), digitized with a MEA2100-System (Multichannel Systems) and recorded using Multi Channel Experimenter software. To investigate acute effects of high glucose, we added either 30 mM D-glucose or mannitol as iso-osmotic control to the perfusate in fellow eyes 20 min before recordings were started.
To determine ON-bipolar cell (BPC, also termed PII) responses, combined photoreceptor (PR, also termed fast PIII) and BPC responses were measured at light (530 nm LED light source) intensities from 0.6 to 20 photons μm−2 in the presence of 100 µM BaCl2 (to inhibit K+ channels in Müller glia).30 Corresponding PR responses were recorded in the presence of 100 µM BaCl2 and 40 µM DL-2-Amino-4-phosphonobutyric acid (DL-AP4, to block neurotransmission from photoreceptors to ON-bipolar cells) and subtracted from combined PR+BPC responses to obtain PII responses31 (figure 1A–C). PR response families were recorded at light (530 nm LED light source) intensities from 6 to 1045 photons μm−2 in the presence of 100 µM BaCl2 and 40 µM DL-AP4. The amplification constant A for phototransduction at a light intensity of 6 photons μm−2 was calculated, as previously described.28 32

Isolation of photoreceptor and ON-bipolar cell function by ex vivo ERG. The PR response family as a measure of ex vivo photoreceptor function in this example trace was recorded in the presence of BaCl2 and DL-AP4 with light stimuli (arrows, B). Combined PR and BPC responses were obtained at the same light intensities in the presence of BaCl2 alone (A) and isolated ON-bipolar cell responses were calculated by subtraction of PR from combined PR+BPC waveforms (C). BPC, ON-bipolar cell; DL-AP4, DL-2-Amino-4-phosphonobutyric acid; ERG, electroretinogram; PR, photoreceptor.
Statistical analysis
Data are displayed as arithmetic means±SD. Statistical analysis was performed using Origin software. Differences between two groups with independent values were determined by two-tailed Student’s t-test, for multiple stimulus intensities by two-way ANOVA.
Data and resource availability
No datasets and no applicable resources were generated or analyzed during the current study.
Results
Type 2 diabetes has different effects on outer retinal physiology under in vivo and ex vivo conditions
Long-term diabetes impairs the function and survival of RGCs and amacrine cells,3 5 7–9 but there is currently no consensus on its effects on phototransduction and synaptic transmission of the rod photoreceptors. Here, we first asked: what is the time course of alterations in rod-mediated light signals caused by type 2 diabetes in habitual blood glucose? To answer this question, we compared scotopic in vivo ERG a-waves and b-waves in db/db mice and their age-matched db/+ controls at 3 and 6 months of age. We used isoflurane anesthesia, which in contrast to ketamine/xylazine anesthesia, did not significantly increase blood glucose in C57BL/6J mice (online supplementary figure 1C).33 34
We did not observe reduced in vivo a-wave and b-wave amplitudes in diabetic compared with non-diabetic control mice at 3 months of age, but rather a slight increase of the in vivo a-wave amplitude, similar to that reported by Johnson et al18 (online supplementary figure 2A, figure 2A,B). a-wave and b-wave implicit times were delayed at this age, suggesting beginning photoreceptor and ON-bipolar cell dysfunction (figure 2C,D). Conversely, in 6-month-old diabetic mice, the scotopic in vivo ERG a-wave and b-wave amplitudes were reduced (online supplementary figure 2B, figure 2E,F) and a-wave and b-wave implicit times delayed (figure 2G,H). The amplification constant (A) of phototransduction, a measure of signal amplification in photoreceptors, was determined based on the leading edge of the a-wave, when b-wave intrusion is negligible, as previously described.32 A was not significantly different between 6-month-old control and diabetic mice (n=8–9, p=0.25), indicating that the activation reactions of phototransduction in rods are not affected by diabetes. Interestingly, we found a trend towards a reduced b/a-wave ratio in diabetic animals compared with non-diabetic controls at 3 months of age, which was highly significant at 6 months of age (table 1).
Supplemental material
b/a-wave ratios in diabetic mice at 3 and 6 months of age

Scotopic a-waves and b-waves are reduced in 6-month-old but not 3-month-old type 2 diabetic mice in vivo. The scotopic a-wave (A, E) and b-wave (B, F) amplitudes as a function of flash energy (QV) for db/+ (black squares) and db/db (red circles) mice at 3 (A–B, n=4–8 eyes from 2 to 4 animals, **p<0.01) and 6 (E–F, n=9–10 eyes from 5 animals, ***p<0.001) months of age. a-wave and b-wave implicit times in db/db (black squares) mice compared with db/+ (red circles) mice at either time point (C–D and G–H, **p<0.01, ***p<0.001).
Although in vivo ERG data by us and others suggest that the light-sensitive current and/or synaptic transmission of rods are compromised during long-term hyperglycemia, it is not known whether these changes are related to an altered extracellular environment or permanent molecular/structural changes in rods or rod bipolar cells caused by diabetes. Moreover, systemic effects of anesthesia, such as hyperglycemia, complicate interpretation of the results obtained by in vivo ERG in animal models of diabetes. To determine the effects of chronic diabetes on the intrinsic functional properties of rods and rod bipolar cells in a normoglycemic environment and to avoid systemic complications of anesthesia, we assessed rod phototransduction and synaptic transmission using ex vivo ERG in 6-month-old control and diabetic mice. Surprisingly, in contrast to the in vivo ERG, the amplitudes of the photoreceptor or ON-bipolar cell components were not significantly changed in 6-month-old db/db mice as compared with age-matched db/+ mice (figure 3A,B). These results demonstrate that transduction or transmission of light signals in rods is not adversely affected by long-term type 2 diabetes under the standard physiological environment used in ex vivo experiments.

Ex vivo photoreceptor and ON-bipolar cell function demonstrates no amplitude changes in the diabetic retina. PR (A) and BPC (B) amplitude data for db/+ (black squares) and db/db (red circles) mice in normoglycemic conditions (A–B, n=11 for PR and n=4–6 for BPC). Averaged PR waveforms at 6 photons μm−2 normalized to maximal response amplitude at 1050 photons μm−2 (C), and quantification of the amplification constant (D, n=9, *p<0.05, see text for details). PR (E, n=11, *p<0.05) and BPC (F, n=4–6) implicit times in db/db (red circles) compared with db/+ (black squares) mice. BPC, ON-bipolar cell; PR, photoreceptor.
Although our amplitude analysis of the ex vivo ERG data suggests that diabetes does not alter the light sensitivity of rod photoreceptors in normal glucose, it is possible that the kinetics of phototransduction activation and inactivation reactions are modulated by diabetes. To test this, we determined the gain of the phototransduction activation reactions and time-to-peak (=implicit time) from ex vivo ERG photoreceptor responses to dim flashes. Interestingly, we found that long-term type 2 diabetes increased A (figure 3C,D) and contrary to in vivo ERG findings, no change was seen in the ex vivo photoreceptor or ON-bipolar cell time to peak (figure 3E,F).
Acute hyperglycemia promotes light responses in rod photoreceptors of non-diabetic, but not diabetic (type 2) mouse retinas
Previous clinical studies demonstrating increased b-wave amplitudes following elevation of blood glucose suggest that retinal function may be modulated by extracellular glucose concentration.25 To investigate this, we used the ex vivo ERG method that allows us to precisely control glucose concentrations. We first evaluated whether acute elevation of extracellular glucose in the perfusate altered light evoked activity of the rod photoreceptors or bipolar cells in C57BL6/J retinas. Supplementation with 30 mM glucose compared with mannitol iso-osmotic control increased the maximal PR response amplitude (figure 4A and C). ON-bipolar cell responses were also slightly increased by acute addition of glucose (figure 4B and D). However, photoreceptor/ON-bipolar cell amplitude ratio was not significantly changed between mannitol and glucose supplementation, indicating that the increased BPC amplitude in the presence of added glucose derives from increased photoreceptor responses.
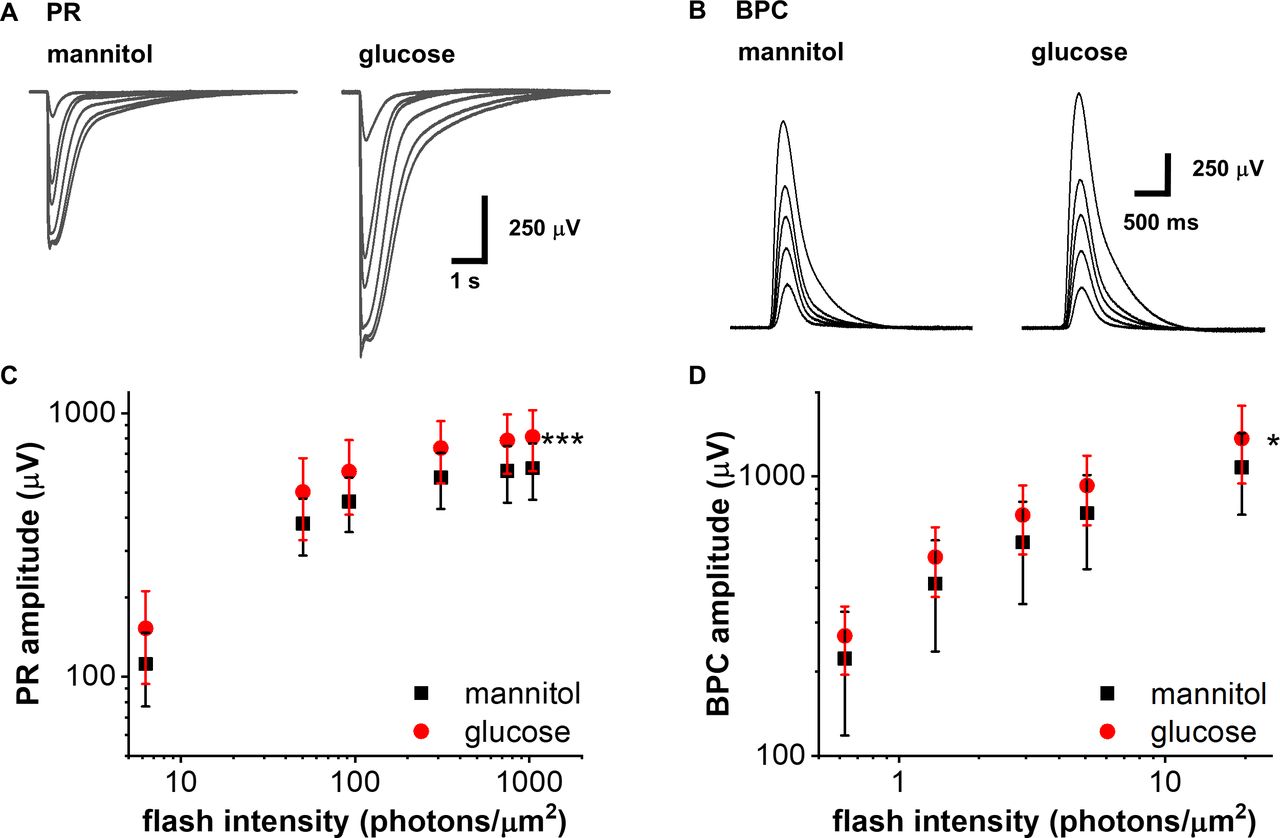
Acute elevation of extracellular glucose increases photoreceptor amplitudes of wild-type mice. Representative ex vivo ERG PR (A) and BPC (B) light response families in normal (iso-osmotic mannitol control, normal glucose+30 mM mannitol) and after supplementation with 30 mM glucose. PR (C, ***p<0.001, n=7) and BPC (D, *p<0.05, n=5) amplitude data in normal (mannitol control, black squares) and high glucose (red circles) in C57BL6/J mice. BPC, ON-bipolar cell; ERG, electroretinogram; PR, photoreceptor.
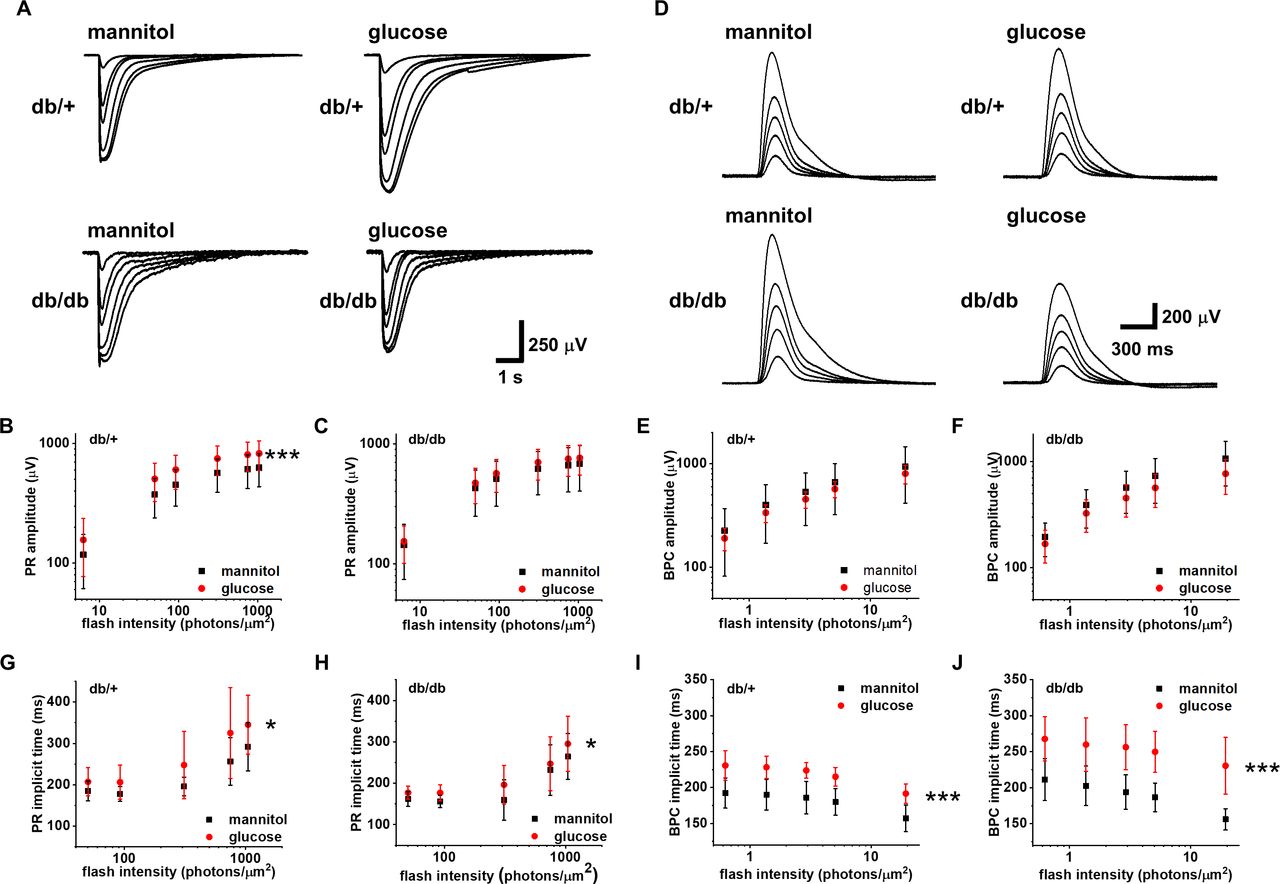
Next, we wanted to know if long-term diabetes alters the acute hyperglycemia-induced augmentation of photoreceptor responses. Diabetes has been shown to cause various long-term adaptations in the retina, including differential expression of glucose transporters.35 Thus, we hypothesized that the photoreceptors in 6-month-old db/db mice are less sensitive to elevated extracellular glucose. Indeed, photoreceptor amplitudes of db/+ control retinas, like C57Bl6 retinas, were significantly augmented in acute hyperglycemia, whereas the photoreceptor responses of db/db retinas were not affected by high glucose exposure (figure 5A–C). On the other hand, we did not observe a significant modulation of the ON-bipolar cell amplitudes by elevated glucose exposure in control or diabetic mice (figure 5D–F). Photoreceptor implicit times were delayed in high glucose compared with mannitol iso-osmotic control in both db/+ and db/db retinas (figure 5G,H). Similarly to photoreceptor implicit times in db/db retinas that were not accelerated compared with db/+ retinas in normal glucose (figure 3E), no significant difference was observed between non-diabetic retinas in normal glucose and diabetic retinas in high glucose (compare black squares in figure 5G and red circles in figure 5H). The amplification constant A as described earlier was not different between high glucose and mannitol treatment in either db/+ or db/db mice (data not shown), suggesting that the increased implicit times of the rod responses must be due to decelerated phototransduction deactivation rather than activation reactions caused by the elevated glucose. Similar to the photoreceptor times to peak, the ON-bipolar cell implicit times lengthened by supplementation with glucose compared with mannitol in both non-diabetic and diabetic animals (figure 5I,J).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Long-term diabetes modulates the response to acute hyperglycemia in photoreceptors. Ex vivo ERG PR (A−C, n=11 retinas) and BPC (D−F, n=6 db/+ and n=4 db/db retinas) light response families and amplitude data in normal (iso-osmotic mannitol control, normal glucose+30 mM mannitol, black squares) and after supplementation with 30 mM glucose (red circles) in 6-month-old control (db/+, ***p<0.001, B, E) and diabetic (db/db, C, F) mice. PR (G, H) and BPC (I, J) implicit times in normal (black squares) and high (red circles) glucose in db/+ (G, I) and db/db (H, J) mice (G−H, *p<0.05, I−J, ***p<0.001). BPC, ON-bipolar cell; ERG, electroretinogram; PR, photoreceptor.
Conclusions
It is widely accepted that diabetes results in loss of RGC and amacrine cells and associated attenuation of oscillatory potentials, both in diabetic animal models and patients.3 7 8 36 Effects of diabetes on photoreceptor and ON-bipolar cell light signaling remain controversial, however. While many studies demonstrated attenuated a-wave and b-wave amplitudes of the scotopic in vivo ERG,9 14 37 38 other reports are contradictory.7 18 39 Causes of these discrepancies remain unclear, but have previously been ascribed to duration of diabetes.9 37 This is supported by our study, which shows reduction of in vivo a-wave and b-wave amplitudes in db/db mice at 6 months of age, whereas the scotopic a-wave was increased in 3-month-old diabetic mice (online supplementary figure 2 and figure 2E,F and 2A,B, respectively). We conclude that changes in in vivo rod photoreceptor function are minor at 3 months of age, but significant dysfunction occurs in uncontrolled type 2 diabetes by 6 months of age. Our data also indicate that signal transmission from rods to rod bipolar cells, measured by the b/a-wave ratio, is impaired in db/db mice, which progresses from 3 to 6 months of age.
Importantly, though, our data imply that altered photoreceptor function in vivo (figure 2) may not be explained by intrinsic dysfunction of the light signal transduction or transmission in diabetic rods (figure 3), but by systemic diabetes-mediated changes that alter rod function. This is consistent with lack of photoreceptor loss19 and absence of reports of reduced scotopic ERGs in patients with diabetes. Since other studies have shown attenuated photoreceptor function in ex vivo transretinal recordings from STZ mice,40 our findings may be model dependent, or restricted to the type 2 diabetic db/db mouse. It is also possible that compromised rod photoreceptor function in the STZ mouse model derives from neurotoxic effects of STZ41 rather than a consequence of diabetes. Our results are consistent with other ex vivo studies showing reduced inhibitory output from amacrine cells but no changes in excitatory output of rods and increased or unaltered ON bipolar cell output in diabetic retinas.10 11 42
The ex vivo ERG technique uniquely allows precise control of the experimental conditions, including extracellular glucose concentration, and isolation of the ERG waveforms from individual retinal cell types. We therefore conclusively show that under normoglycemic conditions and lacking systemic detrimental factors associated with diabetes, generation of light signals and their transmission to rod bipolar cells are not altered by long-term diabetes. What could explain the difference between in vivo and ex vivo ERG in diabetic mice? One obvious difference is the hyperglycemic environment of photoreceptors in diabetic mice in vivo43 compared with normal extracellular glucose in the ex vivo experiments for retinas from both control and diabetic mice. However, increasing the glucose concentration in ex vivo experiments with diabetic mice did not affect their photoreceptor response amplitudes (figure 5A and C). Thus, the difference in the extracellular glucose in vivo cannot explain the decreased in vivo ERG amplitudes.
Other confounding factors include anesthesia-induced systemic effects. Indeed, anesthetic agents are known to affect ERG amplitudes.44 Therefore, one possibility is a different response of retinas in diabetic mice to the systemic changes caused by anesthesia. This is supported by the observation that scotopic a-waves are not altered in conscious human patients with diabetes during in vivo ERG experiments.45 One obvious systemic effect of anesthesia is elevated blood glucose.34 However, previous studies have demonstrated that hyperglycemia in fact promotes retinal signaling in diabetic patients,25 26 whereas light-evoked ex vivo responses from diabetic mouse retinas were unchanged by hyperglycemia in our study. Thus, it is improbable that anesthesia-induced hyperglycemia could explain reduced in vivo ERG a-waves or b-waves in 6-month-old db/db mice. Furthermore, we used isoflurane anesthesia which did not cause anesthesia-induced hyperglycemia. While previous studies concluded that retinal neurodegeneration in db/db mice was abrogated by dietary restriction, it remains unclear whether this was the result of reduced hyperglycemia, body weight or other diabetes-related changes.14 Consequently, we suggest that systemic effects, other than hyperglycemia, that are related to diabetes or general anesthesia affect photoreceptor light responses, resulting in reduced a-wave and b-wave amplitudes in vivo after long-term uncontrolled diabetes in mice.
Several mechanisms by which long-term diabetes reduces the in vivo ERG are plausible, but remain to be further investigated. Deficient retinoid metabolism and consequently decreased rhodopsin content in the diabetic mouse retina have been suggested to contribute to retinal neuronal dysfunction.46 Although increased vascular endothelial growth factor and basic fibroblast growth factor have been correlated with delayed b-waves in patients with diabetic retinopathy,47 effects of inflammatory factors and oxidative stress, which are widely known to play a major role in the pathology of diabetic retinopathy, on the electroretinogram remain unknown. In addition, retinal and choroidal malperfusion48 likely reduce oxygenation of the diabetic retina,49 which results in photoreceptor and ON-BPC dysfunction.50 While rhodopsin content, angiogenic and inflammatory factors, and reactive oxygen species in the retina are unlikely to be affected by isolation and perfusion of the retina, our results showing rapid reversibility of attenuated retinal function in the ex vivo ERG are consistent with improved retinal oxygenation.
It is also possible that altered recording geometry due to different size or body lipid composition of diabetic animals affects in vivo ERG amplitudes. This, however, is not a probable explanation since altered in vivo ERG a-waves and b-waves have been reported both in type 1 (non-obese) and 2 (obese) diabetic animal models. Overall, we conclude that photoreceptor and ON-bipolar cells in diabetic mouse retinas have the potential to mediate normal light signals, although they may have been negatively impacted by altered extracellular environment in diabetes in vivo.
While photoreceptors are highly metabolically active and mostly depend on glucose as their source of energy,51 effects of acute hyperglycemia on retinal function remain unclear, although previous reports suggest a correlation between bipolar cell function and blood glucose levels. In clinical studies, the scotopic b-wave amplitude was increased in hyperglycemic patients with diabetes.25 Similarly, the scotopic a-waves and b-waves were reported to be increased in diabetic ZDF rats, while acute insulin treatment resulted in decreased a-wave amplitudes.18 These results seem at odds with our observation that acutely elevated glucose increased photoreceptor responses in control but not in 6-month-old db/db mice. However, in human patients, glucose levels are typically controlled by insulin, suggesting that the ability to promote photoreceptor responses is lost specifically due to a long-term exposure to an uncontrolled hyperglycemic environment. The difference of our data in comparison with that of Johnson et al. remain unclear, but may be related to different animal models and longer duration of diabetes in our study.
While our experiments did not identify the mechanisms by which light-evoked photoreceptor responses are increased in the presence of additional glucose, likely targets include energy-dependent processes in photoreceptors such as Na+/K+ ATPases, Na+/Ca2+/K+ exchangers and Ca2+ ATPases,51 ATP-sensitive K+ channels expressed in photoreceptors52 or other ATP-dependent processes. The main glucose transporters expressed in photoreceptors are the insulin-dependent GLUT-4 and insulin-independent GLUT-1 which has previously been reported to be reduced in diabetes,35 51 53 54 thus potentially explaining the inability of the photoreceptors in db/db mice to use increased glucose concentrations to promote their light responses. However, these findings are not universal and increased expression of GLUT-1 in diabetes has also been reported.43 Determining underlying causes for the lack of increased photoreceptor function during acute hyperglycemia in diabetic animals was beyond the scope of this study and should be subject to future investigations.
In summary, we demonstrate that rods have the potential to transduce and transmit light signals normally in db/db mice at least up to 6 month of age under a standard ex vivo environment. Differences between diabetic and non-diabetic mice in the in vivo ERG are likely due to systemic factors, which remain to be identified.
References
Footnotes
Presented at Parts of this study were presented in abstract form at the Association for Research in Vision and Ophthalmology Annual Meeting in Vancouver, Canada, 28 April – 2 May 2019.
Contributors SB designed and conducted experiments and wrote the manuscript. LSC contributed to data interpretation and manuscript preparation, FV contributed conceptually to research, designed experiments and wrote the manuscript. SB is the guarantor of this work, had full access to the data and takes responsibility for the integrity and accuracy of the data and its analysis.
Funding This work was supported by National Eye Institute grant EY026651, Research to Prevent Blindness/Dr. H. James and Carole Free Career Development Award and the Diabetes Research Connection (FV), and by an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology and Visual Sciences at the University of Utah.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Institutional Animal Care and Use Committee (IACUC, protocol no. 17-08001) at the University of Utah.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Data are available upon reasonable request.