Article Text

Abstract
Introduction The PIONEER 7 trial demonstrated superior glycemic control and weight loss with once-daily oral semaglutide with flexible dose adjustment versus sitagliptin 100 mg in type 2 diabetes. This 52-week extension evaluated long-term oral semaglutide treatment and switching from sitagliptin to oral semaglutide.
Research design and methods A 52-week, open-label extension commenced after the 52-week main phase. Patients on oral semaglutide in the main phase continued treatment (n=184; durability part); those on sitagliptin were rerandomized to continued sitagliptin (n=98) or oral semaglutide (n=100; initiated at 3 mg) (switch part). Oral semaglutide was dose-adjusted (3, 7, or 14 mg) every 8 weeks based on glycated hemoglobin (HbA1c) (target <7.0% (<53 mmol/mol)) and tolerability. Secondary endpoints (no primary) included changes in HbA1c and body weight.
Results In the durability part, mean (SD) changes in HbA1c and body weight from week 0 were –1.5% (0.8) and –1.3% (1.0) and –2.8 kg (3.8) and –3.7 kg (5.2) at weeks 52 and 104, respectively. In the switch part, mean changes in HbA1c from week 52 to week 104 were –0.2% for oral semaglutide and 0.1% for sitagliptin (difference –0.3% (95% CI –0.6 to 0.0); p=0.0791 (superiority not confirmed)). More patients achieved HbA1c <7.0% with oral semaglutide (52.6%) than sitagliptin (28.6%; p=0.0011) and fewer received rescue medication (9% vs 23.5%). Respective mean changes in body weight were –2.4 kg and –0.9 kg (difference –1.5 kg (95% CI –2.8 to –0.1); p=0.0321). Gastrointestinal adverse events were the most commonly reported with oral semaglutide.
Conclusions Long-term oral semaglutide with flexible dose adjustment maintained HbA1c reductions, with additional body weight reductions, and was well tolerated. Switching from sitagliptin to flexibly dosed oral semaglutide maintained HbA1c reductions, helped more patients achieve HbA1c targets with less use of additional glucose-lowering medication, and offers the potential for additional reductions in body weight.
Trial registration number NCT02849080.
- glucagon-like peptide 1
- dipeptidyl peptidase 4
- treatment outcome
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Oral semaglutide is the first glucagon-like peptide-1 receptor agonist approved for oral administration for type 2 diabetes.
In the PIONEER 7 study, oral semaglutide with flexible dose adjustment provided superior glycemic control and weight loss compared with once-daily sitagliptin 100 mg.
What are the new findings?
This extension phase of the PIONEER 7 study represents the longest follow-up with oral semaglutide to date (2 years), and demonstrated that continued treatment with oral semaglutide with flexible dose adjustment maintained improvements in glycemic control with further weight loss and was generally well tolerated with no new safety signals.
Switching from sitagliptin to oral semaglutide with flexible dose adjustment maintained glycated hemoglobin (HbA1c) reductions with less need for additional glucose-lowering medications, improved the likelihood of achieving an HbA1c target of <7.0%, and may provide additional reductions in body weight.
How might these results change the focus of research or clinical practice?
These results support the long-term use of oral semaglutide in clinical practice in terms of durability of effect and safety, and provide evidence of additional benefits of switching from the dipeptidyl peptidase-4 inhibitor, sitagliptin, to oral semaglutide.
Introduction
Oral semaglutide is the first glucagon-like peptide-1 receptor agonist (GLP-1RA) formulated for oral administration and approved for the treatment of type 2 diabetes (T2D). The efficacy and safety of oral semaglutide in patients with T2D were demonstrated in the global PIONEER phase 3a program, in which oral semaglutide was reported to reduce glycated hemoglobin (HbA1c) and body weight compared with placebo and several commonly used glucose-lowering drugs.1–10
Most PIONEER trials assessed oral semaglutide at fixed once-daily doses of 3, 7, or 14 mg, with initial dose escalation. However, in clinical practice, doses may be adjusted, and in the PIONEER 7 trial oral semaglutide was flexibly dosed (3, 7, or 14 mg once daily) on the basis of efficacy (HbA1c target <7.0% (<53 mmol/mol)) and tolerability criteria. During the 52-week main phase of the trial, once-daily oral semaglutide with flexible dose adjustment provided superior glycemic control and weight loss compared with the dipeptidyl peptidase 4 (DPP-4) inhibitor, sitagliptin (100 mg once daily).7 GLP-1RAs stimulate GLP-1 receptors and mimic the effects of GLP-1, while DPP-4 inhibitors potentiate the effects of endogenously secreted GLP-1 by preventing its degradation.11
The extension phase of PIONEER 7, reported here, had two aims: to evaluate the long-term efficacy and safety of oral semaglutide with flexible dose adjustment; and to evaluate the efficacy and safety when switching from sitagliptin to oral semaglutide with flexible dose adjustment compared with continued sitagliptin.
Materials and methods
Trial design
As reported previously, patients in the main phase of PIONEER 7 received either once-daily oral semaglutide with flexible dose adjustment or once-daily sitagliptin 100 mg as add-on to their glucose-lowering background medication.7 The 52-week, open-label extension phase (with additional 5-week follow-up) commenced immediately thereafter (online supplemental figure 1), from March 28, 2018 to March 27, 2019, at 71 sites in nine countries (Argentina, Austria, Belgium, Egypt, Norway, South Korea, Switzerland, Turkey, and USA).
Supplemental material
The extension included two parallel parts, one of which assessed the durability of treatment effect and the other the effect of switching treatment. In the durability part, patients randomized to oral semaglutide during the main phase who were still on study drug at the start of the extension, and consented to continue, remained on oral semaglutide. The durability part covered the entire treatment period of the trial (0–104 weeks). In the switch part, patients randomized to sitagliptin during the main phase who were still on study drug, and consented to continue, were rerandomized to oral semaglutide or sitagliptin. The switch part covered the extension phase only (52–104 weeks).
In the main phase,7 the primary treatment policy estimand assessed the treatment effect for all randomized patients regardless of treatment discontinuation or use of rescue medication. The secondary trial product estimand assessed the treatment effect for all randomized patients under the assumption that all patients remained on-treatment for the entire duration of the trial and did not use rescue medication.12 These estimands were also used for the switch part of the extension but did not apply to the durability part (see online supplemental material).
Patients
In the main phase, eligible patients were aged ≥18 years (≥19 years in South Korea), had been diagnosed with T2D for ≥90 days before screening, had HbA1c of 7.5%–9.5% (58–80 mmol/mol), were receiving stable daily doses of one to two oral glucose-lowering drugs (metformin, sulfonylureas, sodium-glucose co-transporter 2 inhibitors, or thiazolidinediones) for ≥90 days, and were judged by the investigator to be suitable for a recommended HbA1c target of <7.0% (<53 mmol/mol).7 For the extension, patients were required to still be on randomized treatment (regardless of use of rescue medication and HbA1c level) and to have provided written informed consent to continue in the trial.
Procedures
Patients who remained on oral semaglutide in the durability part continued to have their dose adjusted every 8 weeks using the same clinical criteria as in the main phase. If HbA1c was <7.0% (<53 mmol/mol), dose was unchanged; if HbA1c was ≥7.0% (≥53 mmol/mol), dose was increased (to a maximum of 14 mg), unless moderate-to-severe nausea or vomiting for ≥3 days had been reported in the week before, in which case the dose could be maintained or decreased (to a minimum of 3 mg) at the investigator’s discretion. Patients randomized to sitagliptin in the main phase who continued into the switch part were rerandomized (1:1) at week 52 to once-daily oral semaglutide (with flexible dose adjustment to 3, 7, or 14 mg as per the criteria outlined above; initiated at 3 mg immediately without sitagliptin washout) or to continued once-daily sitagliptin 100 mg. Randomization was conducted using an interactive web response system and stratified based on achievement of HbA1c <7.0% (<53 mmol/mol) and use of rescue medication at week 52.
Patients were instructed to take oral semaglutide once daily with up to half a glass of water (~120 mL/4 fluid oz) in the morning in a fasting state and ≥30 min before the first meal of the day and any other oral medication. Sitagliptin 100 mg was taken once daily with or without food without any dose escalation or adjustment. Rescue medication (ie, intensification of existing glucose-lowering medication and/or initiation of new glucose-lowering medication) used at completion of the main phase was continued at week 52 and was considered background therapy in the extension. Patients continued background glucose-lowering medication as per the start of the extension phase, unless they required additional glucose-lowering medication. In the switch part, rescue medication was only permitted (as per the specified criteria7) after dose escalation in those switching from sitagliptin to oral semaglutide. Patients who prematurely discontinued the study drug were switched to another glucose-lowering medication, including those not allowed as rescue medication, at the investigator’s discretion. Patients were followed up throughout the trial, regardless of rescue medication use or premature discontinuation of study drug, unless consent was withdrawn.
Outcomes
There was no primary endpoint for either part of the extension. Secondary endpoints assessed for the durability part included the following (none of which were confirmatory): changes from baseline (week 0) in HbA1c and body weight at week 104; and whether patients achieved HbA1c <7.0% (<53 mmol/mol), and the composite outcome of HbA1c <7.0% (<53 mmol/mol) or ≥1% HbA1c reduction at week 104. Secondary endpoints assessed for the switch part included the following: changes from week 52 to week 104 in HbA1c and body weight (both confirmatory endpoints), fasting plasma glucose (FPG), and Diabetes Treatment Satisfaction Questionnaire (DTSQ) score; and whether patients achieved HbA1c targets of <7.0% (<53 mmol/mol) and ≤6.5% (≤48 mmol/mol), weight loss ≥5% (since week 52), and two composite outcomes (HbA1c <7.0% (<53 mmol/mol) without hypoglycemia (severe or confirmed by blood glucose concentration <3.1 mmol/L (<56 mg/dL) symptomatic hypoglycemia) and without weight gain since week 52, and HbA1c <7.0% (<53 mmol/mol) and without the need for rescue medication since week 52) at week 104. A post-hoc descriptive analysis of the switch part investigated change from week 52 to week 104 in HbA1c among patients with HbA1c ≥7.5% (≥58 mmol/mol) and ≥8.0% (≥64 mmol/mol) at week 52.
Safety endpoints (assessed from week 0 to week 104 for the durability part and from week 52 to week 104 for the switch part) included the following: adverse events (AEs) and symptomatic hypoglycemic episodes that were severe or confirmed by blood glucose concentration <3.1 mmol/L (<56 mg/dL); and changes in laboratory parameters, vital signs, ECG, and physical examinations at week 104. An independent external event adjudication committee (EAC) validated selected AEs.
Statistical analysis
There were four analysis sets: the durability full and safety analysis sets, and the switch full and safety analysis sets. The full analysis sets were used for efficacy evaluations and comprised all randomized patients; patients contributed to the treatment group based on the study drug to which they were randomized. The safety analysis sets were used for safety evaluations and comprised all randomized patients who had received ≥1 dose of the study drug (during the main phase for the durability part and during the extension phase for the switch part); patients contributed to the treatment group based on the study drug they actually received for the majority of the on-treatment observation period (the duration receiving study drug).
Efficacy endpoints for the durability part were summarized descriptively for the in-trial (duration within trial regardless of rescue medication use or study drug discontinuation) and on-treatment without rescue observation periods (duration receiving the study drug before initiation of rescue medication). For the switch part, a hierarchical closed-testing strategy was used to control the overall type 1 error for the confirmation of efficacy of oral semaglutide when assessed by use of the treatment policy estimand, with superiority for HbA1c required to be demonstrated before testing for superiority for body weight. Under the assumption that 190 patients entered the extension phase, there would be a conditional power of 44% to confirm superiority of oral semaglutide versus sitagliptin for change in HbA1c from week 52 to week 104. The treatment policy estimand was estimated by a pattern mixture model using multiple imputation to handle missing week 104 data. Data collected at week 104, irrespective of premature discontinuation of the study drug or initiation of rescue medication, were included in the statistical analysis. Missing data were imputed within groups defined by study drug and treatment status at week 104, assuming that the likely values of the missing data were best described by observed responses from patients taking the same study drug and with the same treatment discontinuation status and/or rescue medication use. Both the imputation and analysis were based on analysis of covariance models, with region and stratification factors as fixed effects and week 52 measurement as a covariate. Results were combined using Rubin’s rule.13 The trial product estimand was estimated by a mixed model for repeated measurements, with treatment, region, and stratification factors as categorical fixed effects and week 52 measurement as a covariate, all nested within visit. All data collected at scheduled visits prior to premature study drug discontinuation or initiation of rescue medication were included in the statistical analysis. An unstructured covariance matrix for endpoint measurements within the same patient was used.
Safety endpoints were primarily evaluated for the on-treatment period. Deaths and AEs with potentially long latency between onset and diagnosis (cardiovascular disorders, neoplasms, diabetic retinopathy, and rare events) were evaluated for the in-trial period. Further details of the statistical analyses are provided in the online supplemental material.
Results
Durability
There were 253 patients randomized to oral semaglutide at the start of the main phase (week 0) and included in the durability full analysis and safety analysis sets; of these, 185 entered the durability part of the extension phase (week 52) (online supplemental figure 2). In total, 177 patients (70.0%) completed the treatment and 182 patients (71.9%) completed the trial. At baseline (week 0), the mean (SD) HbA1c was 8.3% (0.6%), the mean body weight was 88.9 kg (19.6), and the mean diabetes duration was 8.6 years (6.3) (table 1). Most patients (57.7%) were receiving two types of concomitant glucose-lowering drugs.
Baseline demographics and disease characteristics
Of the 184 patients exposed to the study drug during the extension, 8.7% were on 3 mg, 27.7% on 7 mg, and 63.6% on 14 mg at week 52; respective proportions for the 177 patients still receiving oral semaglutide at week 104 were 6.8%, 19.2%, and 74.0% (online supplemental figure 3). Between weeks 0 and 104, 9.9% of patients received rescue medication (online supplemental table 1).
Efficacy
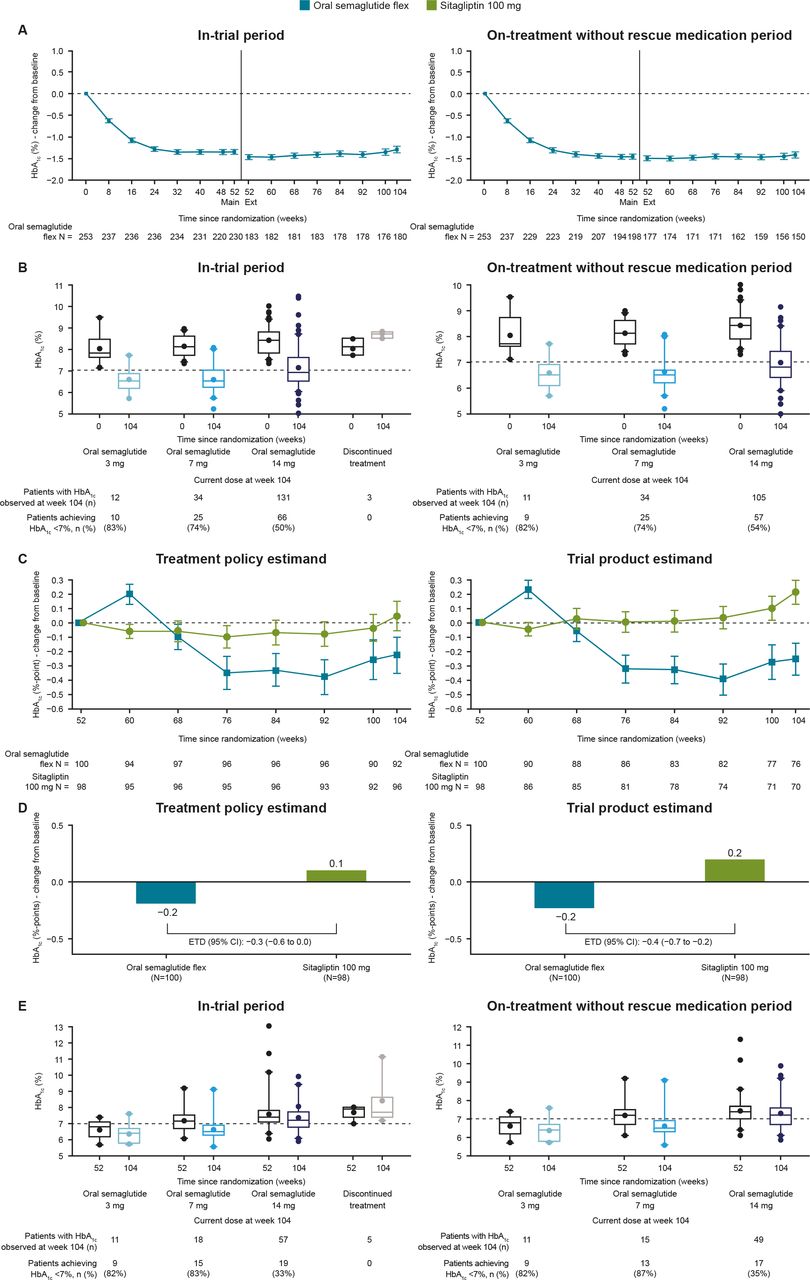
For the in-trial period, the reduction in HbA1c achieved with oral semaglutide in the main phase was maintained for the duration of the extension (figure 1A). Among the patients in the extension, the observed mean (SD) change from baseline (week 0) in HbA1cwas –1.5% (0.8) (–16 mmol/mol (8)) at week 52, and –1.3% (1.0) (–14 mmol/mol (11)) at week 104. The observed changes from baseline (week 0) were similar for the on-treatment without rescue period (mean (SD) –1.5% (0.7) (–16 mmol/mol (8)) at week 52; –1.4%(0.8) (–16 mmol/mol (9)) at week 104).

Glycemic control-related efficacy endpoints. (A) Observed mean change from baseline in HbA1c over the entire trial for the durability part; (B) observed HbA1c at weeks 0 and 104 by current dose at week 104 for the durability part; (C) observed mean change from baseline in HbA1c from week 52 by week for the switch part; (D) estimated change from week 52 in HbA1c at week 104 for the switch part; (E) observed HbA1c at weeks 52 and 104 by current dose at week 104 for the switch part. The durability part includes all patients randomized to oral semaglutide at baseline (week 0). The switch part includes all patients previously randomized to sitagliptin at baseline (week 0) and rerandomized to either oral semaglutide or sitagliptin at the start of the extension phase (week 52). n: number of patients contributing to the mean. Error bars (A and B) represent SEM. ETD, estimated treatment difference; flex, flexible dosing; HbA1c, glycated hemoglobin.
For the in-trial period, body weight was reduced from baseline with oral semaglutide in the main phase, followed by an additional reduction during the extension phase (figure 2A). Among the patients in the extension phase, the observed mean (SD) change from baseline in body weight was –2.8 kg (3.8) at week 52 and –3.7 kg (5.2) at week 104. The observed changes from baseline were similar for the on-treatment without rescue period (mean (SD) –2.8 kg (3.9) at week 52; –3.9 kg (5.2) at week 104).

{kind=link}
{kind=link}
Body weight-related efficacy endpoints. (A) Observed mean change from baseline in body weight over the entire trial for the durability part; (B) observed mean change from baseline in body weight from week 52 by week for the switch part; (C) estimated change from week 52 in body weight at week 104 for the switch part. The durability part includes all patients randomized to oral semaglutide at baseline (week 0). The switch part includes all patients previously randomized to sitagliptin at baseline (week 0) and rerandomized to either oral semaglutide or sitagliptin at the start of the extension phase (week 52). Error bars (A and B) represent SEM. ETD, estimated treatment difference; flex, flexible dosing; n, number of patients contributing to the mean.
The proportion of patients achieving HbA1c <7.0% (<53 mmol/mol) was 62.8% and 56.1% at weeks 52 and 104, respectively, and at week 104, 70.0% achieved the composite outcome of HbA1c <7.0% (<53 mmol/mol) or ≥1% HbA1c reduction (in-trial period data; online supplemental table 2). As expected from the dose adjustment criteria, the majority of patients receiving oral semaglutide 3 or 7 mg at the end of the durability part achieved HbA1c <7.0%; those requiring escalation to oral semaglutide 14 mg appeared to have higher baseline HbA1c values and fewer achieved HbA1c <7.0% (figure 1B). Online supplemental table 2 reports additional supportive endpoints.
Safety
From week 0 to week 104, AEs were reported by 85.0% of patients while on oral semaglutide, most of which were non-serious and of mild or moderate severity (table 2, online supplemental table 3). Gastrointestinal disorders were reported most frequently, especially nausea and diarrhea. Most patients reported their first AE during the initial months of the trial, which correlated with the gradual individualized dose escalation every second month. After week 32, the increase in number of events per patient was generally constant over time.
On-treatment adverse events
Premature discontinuation of oral semaglutide occurred in 9.1% of patients during the trial; most AEs leading to premature discontinuation had onset early in the trial and only one occurred in the extension phase (online supplemental table 4). Blood glucose-confirmed symptomatic hypoglycemic episodes were reported in 7.1% of patients, and the episodes occurred throughout the trial, with the majority occurring in patients receiving sulfonylureas (online supplemental table 5). No severe hypoglycemic episodes were reported.
There were no deaths in the durability part (main and extension phases). EAC-confirmed events are summarized in online supplemental tables 6 and 7. Twelve EAC-confirmed malignant neoplasm events were reported in 11 patients (none thyroid-related and no clustering to specific organ systems). Eight events occurred in the main phase, one at the end of the main phase/start of the extension (week 52) and three during the extension, resulting in an overall event rate of 2.6 per 100 patient-years.
A Medical Dictionary for Regulatory Activities search identified 16 events of diabetic retinopathy or related complications in 13 patients during the trial (online supplemental table 8). All were non-serious and of mild to moderate severity, and most were identified through routine examination. Three had onset during the main phase, four at the start of the extension, and nine during the extension. There were no clinically relevant changes in blood pressure, pulse rate, or renal function (online supplemental table 9).
Switch
Of the 251 patients randomized to sitagliptin in the main phase, 198 were rerandomized in the extension to oral semaglutide (n=100) or sitagliptin (n=98) (switch full analysis set); of these, 197 were exposed to study drug (switch safety analysis set) (online supplemental figure 2). In total, 183 patients (92.4%) completed the treatment and 197 patients (99.5%) completed the trial. Baseline (week 52) characteristics were generally similar between treatment groups, with a mean (SD) HbA1c of 7.4% (1.0%), mean body weight of 86.4 kg (18.0), and mean diabetes duration of 8.8 years (5.9) (table 1). Over half of the patients (62.1%) had HbA1c ≤7.5% at baseline and only 9.6% had HbA1c >8.5% (table 1).
Of the 88 patients treated with oral semaglutide at week 104, 12.5% were on 3 mg, 20.5% on 7 mg, and 65.9% on 14 mg (online supplemental figure 3). Between weeks 52 and 104, 9.0% of patients on oral semaglutide and 23.5% of patients on sitagliptin received rescue medication (online supplemental table 1).
Efficacy
For patients switching from sitagliptin to oral semaglutide, there was an initial slight increase in HbA1c between weeks 52 and 60; after week 60, HbA1c began to decrease until stabilizing at week 76 at a slightly lower level compared with week 52 (figure 1C). For patients continuing sitagliptin, HbA1c was stable throughout the extension, with no further reduction in HbA1c obtained following the main phase. The estimated mean change from week 52 in HbA1c at week 104 for the treatment policy estimand (treatment effect for all randomized patients regardless of treatment discontinuation or use of rescue medication) was –0.2% (–2 mmol/mol) for oral semaglutide and 0.1% (1 mmol/mol) for sitagliptin, with an estimated treatment difference (ETD) (95% CI) of –0.3% (–0.6 to 0.0) (–3 mmol/mol (–7 to 0)) (p=0.0791). Superiority of oral semaglutide over sitagliptin was not confirmed. For the trial product estimand (treatment effect for all randomized patients on-treatment without use of rescue medication), reductions in HbA1c were statistically significantly greater with oral semaglutide versus sitagliptin (ETD (95% CI) –0.4% (–0.7 to –0.2) (–5 mmol/mol (–7 to –2)); p=0.0021) (figure 1D). Within the post-hoc description of the subgroup of patients with HbA1c ≥7.5% at week 52, the observed mean (SD) change from week 52 to week 104 was –0.6% (1.5) with oral semaglutide (n=40) and –0.3% (1.1) with sitagliptin (n=43); respective values for the subgroup with HbA1c ≥8.0% at week 52 were –1.0% (1.2; n=16) and –0.3% (0.8; n=25) at week 104 (in-trial data).
During the extension phase, reductions in body weight that occurred during the main phase continued in both treatment groups, but to a greater extent with oral semaglutide (figure 2B). Estimated mean change in body weight from week 52 to week 104 for the treatment policy estimand was –2.4 kg for oral semaglutide and –0.9 kg for sitagliptin (ETD (95% CI) –1.5 kg (–2.8 to –0.1); p=0.0321). Superiority of oral semaglutide over sitagliptin was not tested (as superiority was not confirmed for the change in HbA1c). For the trial product estimand, reductions in body weight were statistically significantly greater with oral semaglutide versus sitagliptin (ETD (95% CI) –1.8 kg (–3.3 to –0.3); p=0.0176) (figure 2C).
More patients achieved the HbA1c targets of <7.0% and ≤6.5%, body weight loss of ≥5%, and the composite outcome of HbA1c <7.0% (<53 mmol/mol) without severe or blood glucose-confirmed symptomatic hypoglycemia and without weight gain, with oral semaglutide than sitagliptin for the treatment policy estimand; the odds of achieving these outcomes were significantly greater with oral semaglutide versus sitagliptin (table 3). There were similar results for the trial product estimand (table 3). Similar to the durability part, most patients receiving oral semaglutide 3 or 7 mg at week 104 achieved HbA1c <7.0%; those requiring escalation to oral semaglutide 14 mg appeared to have higher baseline HbA1c and fewer achieved HbA1c <7.0% (figure 1E). Reductions in FPG were not significantly different with oral semaglutide versus sitagliptin for the treatment policy estimand, but were significantly greater with oral semaglutide for the trial product estimand (table 3).
Supportive endpoints at week 104 for the switch part*
For the treatment policy estimand, from week 52 to week 104 there was no significant difference in change in total DTSQ treatment satisfaction score (or any individual DTSQ item) for oral semaglutide versus sitagliptin. For the trial product estimand, the ‘satisfaction with treatment’ item was significantly improved with oral semaglutide versus sitagliptin, and other items and total treatment score were similar between groups (online supplemental figure 4). Online supplemental table 10 reports additional supportive endpoints.
Safety
In the switch part of the extension (from week 52 to week 104), safety findings were similar to the main phase, in that more patients reported AEs with oral semaglutide than sitagliptin (driven by the greater incidence of gastrointestinal AEs), and most AEs were non-serious and of mild or moderate severity (table 2, online supplemental table 3). As in the main phase, gastrointestinal disorders, such as nausea, diarrhea, and vomiting, were the most frequently reported events with oral semaglutide. Six patients prematurely discontinued the study drug due to AEs with oral semaglutide (none in the continued sitagliptin group), with most of these AEs (8 of 10) occurring within the first 8 weeks of switching (online supplemental table 4).
No patients experienced severe hypoglycemic episodes. The incidence of blood glucose-confirmed symptomatic hypoglycemic episodes was low and similar in both treatment groups (online supplemental table 5). Most episodes occurred in patients taking background sulfonylurea (1 of 2 for oral semaglutide; 10 of 12 for sitagliptin).
There were no deaths during the switch part. Two EAC-confirmed malignant neoplasm events (one kidney and one lung) occurred in two patients receiving oral semaglutide, with an event rate of 1.8 per 100 patient-years (online supplemental table 7). Diabetic retinopathy or related complications were reported in four patients receiving oral semaglutide and in two patients receiving sitagliptin during the switch part; all were non-serious and of mild-to-moderate severity, with most identified through routine examinations (conducted at weeks 52 and 104) (online supplemental table 8). There were no clinically relevant changes in blood pressure, pulse rate, or renal function (online supplemental table 11).
Discussion
Treatment guidelines for T2D highlight the need for an individualized approach,14 15 and in clinical practice doses of therapeutic agents are frequently adjusted to achieve optimum results. The main phase of the PIONEER 7 study demonstrated that oral semaglutide dosing can be individualized, and that it is able to provide greater benefits to patients than sitagliptin.7 This long-term, open-label extension builds on these results, with the durability part representing the longest follow-up reported to date for oral semaglutide. Continued flexible dosing of oral semaglutide maintained reductions in HbA1c and rates of achievement of HbA1c goals, and resulted in further reductions in body weight over 104 weeks, while remaining well tolerated with no new safety signals. As expected for a GLP-1RA,16 17 and consistent with other trials from the PIONEER program,1–5 7–10 gastrointestinal events were the most frequently reported AEs with oral semaglutide and the AEs that most often resulted in premature discontinuation. However, the majority of these AEs occurred in the early part of the main phase, and only one patient discontinued due to an AE in the second year of the durability part, indicating that discontinuations due to gastrointestinal AEs with oral semaglutide are likely to occur early on in treatment (during the initial dose-escalation phase) and not with long-term use.
The switch part of the extension investigated the effect of switching from sitagliptin to oral semaglutide with flexible dose adjustment in a population with relatively well-controlled T2D. In the extension, after switching, an initial slight increase in HbA1c was observed due to the withdrawal of sitagliptin and the gradual dose-escalation regimen for oral semaglutide, with patients initiating oral semaglutide at 3 mg (the lowest approved starting dose for the dose-escalation strategy). HbA1c decreased once patients could be escalated to oral semaglutide 7 and 14 mg (ie, at weeks 60 and 68), stabilizing at week 76. By week 104, there were small reductions in HbA1c with oral semaglutide (–0.2%) compared with a small increase in the sitagliptin group (0.1%). Difference between treatments was not significant by the treatment policy estimand, although reached significance by the trial product estimand (on-treatment without use of rescue medication), which may reflect the greater use of rescue medication in the sitagliptin group (24% vs 9% of patients in the oral semaglutide group). The difference in HbA1c, although not significant in terms of absolute changes for the treatment policy estimand, may still have had an impact on goal achievement due to the baseline HbA1c levels when switching (mean HbA1c of 7.4%). Patients switched to oral semaglutide were more likely to achieve HbA1c targets of <7.0% (<53 mmol/mol) than patients who continued on sitagliptin.
Many patients had HbA1c approaching or below the treatment target at the start of the switch part (week 52), with HbA1c ≤7.5% in nearly two-thirds of patients. This may have limited the potential for further HbA1c reductions with oral semaglutide, given the trial was designed for the achievement of glycemic control (HbA1c <7.0%),7 not to maximize HbA1c reduction, and as the flexible dose regimen for oral semaglutide required HbA1c to be ≥7.0% for dose escalation. However, despite this, 30.4% of patients rerandomized to oral semaglutide achieved HbA1c ≤6.5% at week 104, compared with 11.5% of sitagliptin-treated patients. A third of patients who were on-treatment at week 104 were not receiving the highest (14 mg) dose of oral semaglutide. Those patients who required escalation to 14 mg appeared to have a smaller mean decrease in HbA1c; however, it is unknown whether this may have been related to baseline HbA1c status, reduced compliance with treatment, interindividual variability in response to oral semaglutide or some other factor. Among subgroups of patients with higher baseline HbA1c levels, greater HbA1c reductions were seen in both treatment groups; however, this post-hoc assessment was only conducted in a small number of patients and was not analyzed statistically.
Switching from sitagliptin to oral semaglutide appeared to result in body weight reductions by week 104, although superiority was not tested given the hierarchical testing strategy. As in the main phase,7 no significant differences were seen in the switch part between oral semaglutide and sitagliptin for DTSQ total treatment satisfaction score or for the items relating to convenience and flexibility of treatment. This suggests that the dosing conditions for oral semaglutide had no impact on treatment satisfaction when switching from sitagliptin.
More gastrointestinal events (a well-known class effect with GLP-1RAs16 17) occurred in patients switching from sitagliptin to oral semaglutide than in those continuing sitagliptin. A similar finding was observed when switching to another GLP-1RA, liraglutide, from sitagliptin.18 In patients randomized to oral semaglutide with flexible dose adjustment in the switch part, the proportion discontinuing due to AEs (6%) was broadly similar to that observed in those randomized to flexibly dosed oral semaglutide in the main phase of PIONEER 7 (9%).7 While the potential for improved tolerability was part of the rationale for exploring flexible dosing, several other studies of oral semaglutide without flexible dose adjustment report comparable discontinuation rates.1–4 As such, it appears that the individualized and slower approach to dose escalation (every 8 weeks vs every 4 weeks in other oral semaglutide studies) did not impact discontinuation rates; however, as in the main phase, the extension was conducted open-label, which could have influenced the actions of patients and investigators. In the switch part of the study, the majority of AEs leading to oral semaglutide discontinuation occurred within 8 weeks of rerandomization, after patients initiated oral semaglutide 3 mg but before escalation to higher doses. Again, this is consistent with the results of the main phase of PIONEER 7, and suggests that if the initial treatment with the 3 mg dose is tolerated, subsequent dose escalation is also likely to be tolerated.
Overall event rates of malignant neoplasms were similar to those in the much larger PIONEER 6 study, which reported rates of EAC-confirmed malignant neoplasms of 2.5 and 2.9 per 100 patient-years in the oral semaglutide and placebo groups, respectively (median follow-up of 15.9 months).6 The frequency of EAC-confirmed malignant neoplasms over 78 weeks of treatment was also similar with oral semaglutide and sitagliptin in the long-term safety trial, PIONEER 3.3 A meta-analysis including 3446 patients treated with subcutaneous semaglutide for up to 104 weeks and 419 patients treated with oral semaglutide for 26 weeks concluded that there was no increased risk of malignant neoplasms with semaglutide when compared with placebo or other glucose-lowering treatments (OR (95% CI) 0.89 (0.35 to 2.22); p=0.80).19 Consistent with other studies of oral semaglutide,1–10 no clustering of malignancies to specific organ systems was observed.
The key strengths of this study include the flexible nature of the dosing regimen for oral semaglutide, which aids relevance for clinical practice, the long-term follow-up, the high treatment completion and trial completion rates for the extension phase, and the low amount of missing data. Our study also has some limitations, in addition to its open-label nature. First, as highlighted earlier, the relatively low HbA1c at week 52 and dose-escalation criteria for oral semaglutide limited the ability to evaluate the relative HbA1c-lowering efficacy of switching from sitagliptin to oral semaglutide. When designing the extension, the main phase was ongoing and the mean HbA1c levels at the beginning of the extension were not known. Second, the number of patients who continued into the extension phase was relatively small, particularly in the switch part, limiting the ability to assess the incidence of infrequent side effects. This also resulted in a low power for the confirmatory endpoints in the switch part.
In conclusion, this study demonstrates that long-term use of oral semaglutide with flexible dose adjustment results in durable improvements in glycemic control and further reductions in body weight and is generally well tolerated. Switching from sitagliptin to flexible dose-adjusted oral semaglutide maintained HbA1c reductions, with less need for additional treatment intensification, and offers the potential for additional reductions in body weight, but is associated with an increase in gastrointestinal events.
Acknowledgments
The authors would like to thank the patients participating in this trial, the investigators, all trial site staff, and all Novo Nordisk employees involved in the trial. The authors would also like to thank Solomon Nuhoho of Novo Nordisk for reviewing the manuscript, and Andy Bond (Spirit Medical Communications Group) for assistance with medical writing and editorial support, funded by Novo Nordisk A/S.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Collaborators Trial Investigators
Argentina: Claudia Issa, Sanatorio Güemes, Francisco Acuña de Figueroa 1228/1240, CABA; Lucas Rista, CEDyN, Balcarce 637, Rosario; Silvia Gorban de Lapertosa, CUIFC, Sargento Cabral 2001, Corrientes.
Austria: Thomas Pieber, Medizinische Universität Graz, Univ. Klinik für Innere Medizin, Klinisch Abteilung für Endokrinologie und Diabetologie, Auenbruggerplatz 15, Graz; Rudolf Prager, KH Hietzing mit Neurologischem, Zentrum Rosenhügel, 3. Med. Abteilung, Pavillon 4, 2. Stock Wolkersbergenstr. 1, Wien; Evelyn Fließer-Görzer, Ordination Dr. Fließer-Görzer, Kastaniensiedlung 1, St.Stefan.
Belgium: Ann Mertens, UZ Leuven - Campus Gasthuisberg, Department of Endocrinology, Herestraat 49, Leuven; Ides Colin, CHR Hôpital de Warquignies, Department of Endocrinology, Rue de Chauffours 27, Boussu; Vanessa Preumont, Cliniques universitaires St. Luc, Endocrinologie et Nutrition, Avenue Hippocrate, 10, Bruxelles; André Scheen, CHU de Liège - Sart Tilman, Laboratoire de Diabétologie, Tour de Pathologie, 2ème étage, Domaine Universitaire du Sart-Tilman, Avenue de l'Hôpital 1, Liège; Guy T’Sjoen, UZ Gent, Dienst Endocrinologie, Corneel Heymanslaan 10, Gent; Luc Van Gaal, UZ Antwerpen, Dienst Endocrinologie, Diabetologie en, Metabole Ziekten, Wilrijkstraat 10, Edegem; Chris Vercammen, AZ Imelda, Dienst Endocrinologie, Imeldalaan 9, Bonheiden.
Brazil: Freddy Goldberg Eliaschewitz*, CPCLIN - Centro de Pesquisas Clínicas, Rua Goiás, 193, Higienópolis, São Paulo; Luis Henrique Santos Canani*, Centro de Pesquisas em Diabetes Ltda., Rua Gonçalo de Carvalho, 412, Bairro Floresta, Porto Alegre; Jorge Luiz Gross*, Centro de Pesquisas em Diabetes Ltda., Rua Gonçalo de Carvalho, 412, Bairro Floresta, Porto Alegre.
Egypt: Samir Helmy Assaad Khalil, Alexandria CRC, New Hospital building, Faculty of Medicine, Alexandria University, 17 Champollion Street, Messallah, Alexandria; Mohamed Hesham Mohamed Fahmy El Hefnawy, National Institute of Diabetes and Endocrinology, 16 Kasr Al Ainy St., Cairo; Ibrahim Naguib El Ebrashy, Diabetes Outpatient Clinic, Kasr Elaini St. Faculty of Medicine, Cairo University, Cairo; Salah Abo Shelbaya, Diabetes Clinical Research Centre (DCRC), Faculty of Medicine, Ain Shams University, Cairo.
Norway: Hanne Løvdal Gulseth, Aker sykehus, Oslo universitetssykehus HF, Trondheimsveien 235, Oslo; Hans Olav Høivik, M3 Helse, Storhamargata 34, Hamar; John Cooper*, Stavanger Helseforskning, Jan Johnsensgate 5, Stavanger; Cecilie Wium, Lipidklinikken, Oslo Universitetssykehus Rikshospitalet., Forskningsveien 2B, Oslo; Frode Helland, Hallset Legesenter, Selsbakkveien 37, Trondheim.
Republic of Korea: Sei Hyun Baik, Korea University Guro Hospital, 148, Gurodong-ro, Guro-gu, Seoul; Kwan-Woo Lee, Ajou University Hospital, 164, World Cup-ro, Yeongtong-gu, Suwon; Ji A Seo, Korea University Ansan Hospital, 123, Jeokgeum-ro, Danwon-gu; Nan Hee Kim, Korea University Ansan Hospital, 123, Jeokgeum-ro, Danwon-gu; In Joo Kim, Pusan National University Hospital, 179 Gudeok-ro, Seo-gu, Busan; Young Min Cho, Seoul National University Hospital, 101, Daehak-ro, Jongno-gu, Seoul; Eun Seok Kang, Severance Hospital, 50-1 Yonsei-ro, Seodaemun-gu, Seoul; Choon Hee Chung, Wonju Severance Christian Hospital, 20, Ilsan-ro, Wonju, Gangwondo.
Switzerland: Stefan Fischli, Endokrinologie/Diabetologie, Luzerner Kantonsspital, Spitalstrasse 16, Luzern; Alain Golay*, Service d'enseignement thérapeutique pour maladies chroniques, Hôpitaux Universitaires de Genève, Villa Soleillane 7, Chemin Venel, Genève; Cornelia Keller*, Endokrinologie/ Diabetologie, Kantonsspital Winterthur, Brauerstrasse 15, Winterthur; Markus Laimer*, Universitätsklinik für Diabetologie, Endokrinologie und Metabolismus, Inselspital Bern, Freiburgstrasse 4, Bern; Gottfried Rudofsky, Stoffwechselzentrum, Kantonsspital Olten, Fährweg 6, Gebäude M/ Eingang Ost, Olten; Bernd Schultes, eSwiss Medical & Surgical Center, Brauerstr. 97, St. Gallen; Simon Stäuble, MedicoPlus Health Care AG, Spitalstrasse 26a, Einsiedeln; Stefan Bilz, Kantonsspital St. Gallen, Endokrinologie/Diabetologie/Osteologie, Rorschacherstrasse 95, St. Gallen.
Turkey: Aytekin Oğuz, İstanbul Medeniyet Üniversitesi Göztepe EAH, Merdivenköy Polikliniği, Dahiliye ve Diyabet, Poliklinikleri No:21, Kadıköy/İstanbul; Esra Ataoglu, Haseki EAH 3.Blok Kat.2 4., Dahiliye Uzman Odası, Fatih/İstanbul; Dilek Berker, Ankara Numune Hast., C Blok, Kat.3, Endokrin Bölümü, Ankara; Ramazan Sarı, Akdeniz Üni. Tıp Fak., Hastanesi Endokrin ve Metabolizma Polikliniği H, Blok 2. Kat, Konyaaltı/Antalya; Nazire Aladağ, Kartal Eğitim Araştırma Hastanesi, Başhekimlik Binası, 1. Kat, Diyabet Polikliniği, Kartal/İstanbul; Ali Özdemir*, Fatih Sultan Mehmet Eğitim ve Araştırma Hastanesi, Endokrinoloji Bilim Dali, Ataşehir/İstanbul; Tamer Tetiker, Adana Çukurova Üniversitesi, Tıp Fakültesi Endokrinoloji Bilim Dali, Zemin Kat, Balcalı/Adana; Dilek Gogas Yavuz, Fevziçakmak Mah., Muhsin Yazıcıoğlu Cad., Marmara Üni. Pendik EAH., Kat 9, Endokronoloji Klinik Araştırma Odası, Üst, Kaynarca/Pendik; Şafak Akın, Recep Tayyip Erdoğan Üniversitesi Eğitim ve Araştırma Hastanesi, Endokrin Polikliniği, İslampaşa Mahallesi, Şehitler Caddesi No.74, Rize.
USA: Daniel Weiss, Your Diabetes Endocrine Nutrition Group, Inc., 8300 Tyler Blvd, Mentor, Ohio; Leslie Joseph Klaff, Rainier Clinical Research Center Inc., 723 SW 10th Street, Renton, Washington; Jeffrey Geohas, Evanston Premier Healthcare Research, LLC, 2500 Ridge Ave., Evanston, Illinois; Emily J. Morawski, Holston Medical Group, 105 West Stone Drive, Kingsport, Tennessee; Bryce A. Palchick, Preferred Primary Care Physicians, 140 Curry Hollow Rd, Pittsburgh, Pennsylvania; Debra L. Weinstein*, Zasa Clinical Research, 8188 Jog Road, Boynton Beach, Florida; Harold Bays, L-MARC Research Center, 3288 Illinois Avenue, Louisville, Kentucky; John Bernard Buse, University of North Carolina, UNC Diabetes Care Center, 300 Meadowmont Village Circle, Chapel Hill, North Carolina; Belkis Delgado, San Marcus Research Clinic, Inc., 5941 NW 173, Miami, Florida; James C. LaRocque*, Virginia Endocrinology Research, 3205 Churchland Blvd, Chesapeake, Virginia; William Reid Litchfield, Desert Endocrinology Clinical Research Center, 2415 West Horizon Ridge Pkwy, Henderson, Nevada; Ernie Riffer, Clinical Research Advantage, Inc./Central Phoenix Medical Clinic, LLC, 7600 North 15th Street, Phoenix, Arizona; Alexander White, Progressive Medical Research, 5111 Ridgewood Ave., Port Orange, Florida; Stephen Ong, MD Medical Research, Inc., 6357 Oxon Hill Rd, Oxon Hill, Maryland; Narendra A. Godbole, Clinical Research Advantage, Inc./Summit Medical Group Arizona, LLC, 5620 W. Thunderbird Rd, Glendale, Arizona; Louis J. Aronne, Weill Cornell Medical College, Comprehensive Weight Control Program, 1165 York Ave., New York, New York; Dan Alexandru Streja, Infosphere Clinical Studies, Inc., 15243 Vanowen Street, Van Nuys, California; Thomas Michael O'Connor, American Health Network of Indiana, LLC, 300 E Boyd Ave., Greenfield, Indiana; Ahmed A. Arif, AA MRC LLC, 1201 Flushing Road, Flint, Michigan; Bruce Bode*, Atlanta Diabetes Associates, 1800 Howell Mill Road, Atlanta, Georgia; Mary Beth Manning, Rapid Medical Research, Inc., 3619 Park East Drive, Cleveland, Ohio; Kanagaratnam Sivalingam, First Valley Medical Group, 44725 N. 10th Street West, Lancaster, California; Edward W. Braun, Midtown Medical Center, 6919 N. Dale Mabry Hwy, Tampa, Florida; Donald C. Eagerton, Carolina Health Specialists, 945 82nd Parkway, Myrtle Beach, South Carolina; Jeanne Pereles-Ortiz, Billings Clinic Research, 1045 North 30th Street, Billings, Montana; Christopher H. Sorli, Billings Clinic Research, 1045 North 30th Street, Billings, Montana; Michael Winnie, Corpus Christi Family Wellness Center, 5920 Saratoga Blvd, Corpus Christi, Texas; Paul L. Beckett, Elite Clinical Trials, 1443 Parkway Drive, Blackfoot, Idaho; Alexander Vance Murray, PharmQuest, 806 Green Valley Road, Greensboro, North Carolina; Jonathan Condit, American Health Network of Indiana, LLC, 3631 N. Morrison Rd, Muncie, Indiana; Philip R. Nicol, The Diabetes Center, LLC, 11945 Grandhaven Dr, Murrells Inlet, South Carolina; Stephen Aronoff*, Research Institute of Dallas, 10260 N. Central Expressway, Dallas, Texas; Mark L. Warren, Physician's East Endocrinology, 1006 WH Smith Blvd, Greenville, North Carolina; Matthew P. Finneran, Family Practice Center of Wadsworth, Inc., 251 Leatherman Road, Wadsworth, Ohio; Eileen M. Palace*, The Center for Sexual Health, 3500 North Causeway Blvd., Metairie, Louisiana; Samuel N. Lederman, Altus Research, Inc., 4671 S. Congress Ave., Lake Worth, Florida.
*Trial sites that were approved by independent ethics committee/institutional review board and participated in main phase but not extension phase of trial.
Contributors JBB, TRP, CLH, and EC contributed to the trial design. JBB, BWB, AM, YMC, EC, MAN, and TRP contributed to the conduct of the trial and the data collection. EC and MAN contributed to the data analysis. All authors interpreted the data and participated in writing the manuscript, with the support of medical writing services provided by the funder. All authors read and approved the submitted version of the report.
Funding This trial was funded by Novo Nordisk A/S, Denmark. The funder participated in the study design, data collection, analysis and interpretation of the data, and in the writing of the report. The authors provided the final decision to submit the paper for publication. JBB was supported by the US National Institutes of Health (UL1TR002489, P30DK124723).
Competing interests JBB reports contracted consulting fees paid to the University of North Carolina (Chapel Hill, North Carolina, USA) from Adocia, AstraZeneca, Dance Biopharm, Dexcom, Elcelyx Therapeutics, Eli Lilly, Fractyl, GI Dynamics, Intarcia Therapeutics, Lexicon, MannKind, Metavention, NovaTarg, Novo Nordisk, Orexigen, PhaseBio, Sanofi, Senseonics, Shenzhen HighTide, Takeda, vTv Therapeutics, and Zafgen; grant support from AstraZeneca, Eli Lilly, GI Dynamics, GlaxoSmithKline, Intarcia Therapeutics, Johnson & Johnson, Lexicon, Medtronic, Novo Nordisk, Orexigen, Sanofi, Scion NeuroStim, Takeda, Theracos, and vTv Therapeutics; personal fees from Neurimmune AG and Fortress Biotech; and holds stock options in Mellitus Health, PhaseBio, and Stability Health. BWB reports personal fees from Adocia, AstraZeneca, Boehringer Ingelheim, Intarcia, Janssen, Lilly, MannKind, Medtronic, Novo Nordisk, Sanofi, and Senseonics; grant support from Boehringer Ingelheim, Dexcom, Diasome, Janssen, Lilly, MannKind, Medtronic, Novo Nordisk, Sanofi, and Senseonics; and holds shares in Aseko. AM reports board membership and consultancy fees paid to KU Leuven (Leuven, Belgium) from AstraZeneca, Merck Sharp & Dohme, Novo Nordisk, and Sanofi; and payment for lectures from Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly, Johnson & Johnson, Merck Sharp & Dohme, Novartis, Novo Nordisk, and Sanofi. YMC reports grants from AstraZeneca, LG, and Sanofi; and consulting fees from Hanmi. EC, CLH, and MAN are employees of and hold shares in Novo Nordisk. TRP reports board membership and personal fees from Adocia, Arecor, AstraZeneca, Novo Nordisk, and Sanofi; and payment for lectures from Novo Nordisk.
Patient consent for publication Not required.
Ethics approval The trial adhered to ICH Good Clinical Practice and the Declaration of Helsinki, and the study protocol was approved by the Institutional Review Board/Independent Ethics Committee at each study site (online supplemental material). All patients provided written informed consent prior to any trial-related activities in the main phase of the trial, and were required to sign an addendum to the informed consent in order to continue in the extension phase.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Data will be shared with researchers who submit a research proposal approved by an independent review board. Access request proposals can be found at novonordisk-trials.com. Data will be made available after research completion and approval of the product and product use in the EU and the USA. Individual participant data will be shared in data sets in a de-identified and anonymized format. There will not be any limitations on how these data can be used.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.