Article Text

Abstract
Introduction Insulin-like growth factor-1 (IGF-1) has been implicated in fetal and early-life growth and development of type 2 diabetes (T2D). We aimed to examine the interaction between circulating IGF-1 and birth weight in relation to risk of T2D.
Research design and methods We included 181 090 adults, aged 39–70 years in the UK Biobank Study, who were free of diabetes or major cardiovascular diseases at baseline. Serum IGF-1 levels were determined using chemiluminescent immunoassay method. Birth weight was self-reported; a Genetic Risk Score (GRS) was calculated to define the genetically determined birth weight. The outcome was the incidence of T2D.
Results We identified 3299 incident T2D cases over an average of 9.9 years of follow-up. Among the participants with birth weight of ≥2.5 kg, IGF-1 levels were inversely associated with T2D risk in a dose-dependent manner (p-trend<0.001). In contrast, the association was not significant among those with birth weight of <2.5 kg (p-interaction=0.001). The GRS of birth weight did not interact with IGF-1 levels on T2D risk.
Conclusions Our results indicate that birth weight significantly modifies the relation between adulthood levels of circulating IGF-1 and the risk of T2D. Our findings highlight the importance of early-life risk factors in the development of the lifecourse prevention strategies targeting IGF-1 and T2D.
- type 2 diabetes mellitus
- birth weight
- gene–environment Interaction
- epidemiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Lifecourse susceptibility to type 2 diabetes (T2D) is determined jointly by risk factors exposed in both early life and adulthood.
Insulin-like growth factor-1 (IGF-1) has been implicated in fetal and early-life growth and development of T2D.
What are the new findings?
Birth weight significantly modifies the relation between adulthood levels of circulating IGF-1 and the risk of T2D.
Among the participants with birth weight of ≥2.5 kg, IGF-1 levels were inversely associated with T2D risk in a dose-dependent manner.
How might these results change the focus of research or clinical practice?
Early-life risk factors need to be considered for lifecourse prevention and treatment targeting on IGF-1 and T2D.
Introduction
Type 2 diabetes (T2D) is a serious public health threat associated with high morbidity and mortality rates. T2D disables people at their most productive age, impoverishes families and reduces the life expectancy, which places a considerable health and economic burden borne by both individuals and health systems.1 2 Compelling evidence has shown that the lifecourse susceptibility to the development of T2D is determined jointly by risk factors exposed in both early life and adulthood.3–6
Insulin-like growth factor-1 (IGF-1), a hormone predominately produced by the liver when stimulated by growth hormone,7 is similar to insulin in molecular structure and acts synergistically with insulin to maintain glucose homeostasis through binding its own receptors and insulin receptors in different tissues.8–10 Notably, the regulatory effect of IGF-1 on glucose metabolism and development of T2D can be tracked back to the early-life development windows. Prenatal/postnatal deficiency of IGF-1 induced by intrauterine growth restriction may cause ‘diabetic’ aging trajectory toward adulthood, and the decreased levels of IGF-1 during early life may persist into adulthood.11 12 A recent Mendelian randomization study reported a potentially causal relation between the elevated IGF-1 levels and a higher risk of T2D13; however, little is known about whether the association between adulthood IGF-1 levels and T2D risk could be modified by birth weight.
In this present study, we examined the interaction between circulating IGF-1 and birth weight on the risk of T2D in 112 736 women and 68 354 men from the UK Biobank Study. We also investigated whether the association between IGF-1 and T2D could be modified by a Genetic Risk Score (GRS) of birth weight.
Methods
Study population
We used data from the UK Biobank, which is a large population-based prospective cohort study for long-term study of genetic and lifestyle determinants of a wide range of common diseases of middle age and old age. Detailed information on study design has been presented elsewhere.14 Briefly, between 2006 and 2010, a total of 502 506 community-dwelling participants (229 122 men and 273 384 women) aged 37–73 years were recruited from 22 assessment centers across England, Scotland, and Wales. Participants provided information on sociodemographics, habitual diet, lifestyle factors (smoking status, alcohol intake, sleep duration and habitual physical activity), early life exposures (birth weight and maternal smoking) and medical records through touch-screen questionnaires. Physical measurements (anthropometrics) and biological samples (blood, urine and saliva) were also obtained at recruitment.
Laboratory measurements
Blood samples were collected from consenting participants at recruitment and a repeat measurement was conducted among approximately 20 000 participants between 2012 and 2013. Samples were transferred to a dedicated central laboratory, separated by components and stored at UK Biobank (−80°C and LN2) until analysis. Serum IGF-1 concentrations were determined using chemiluminescent immunoassay method (DiaSorin Liason XL). The coefficient of variation for IGF-1 ranged between 5.29% and 6.18%. First repeat measurements of IGF-1 levels were available from 17 699 participants. Total cholesterol (TC), triglyceride (TG), high-density lipoprotein cholesterol (HDL-C), and C reactive protein (CRP) levels were measured using robust and reliable analytical methods on a chemistry analyzer (AU5800 Analyzer, Beckman Coulter). Full details on assay performance have been published previously.15
Assessment of birth weight
All the participants were asked, ‘Do you know your birth weight?’ and they were further asked to enter their own birth weight if they answered ‘yes’. Data on birth weight were available for 293 431 participants. The self-reported birth weight has been validated and demonstrated to be reliable.16
Genetic Risk Score
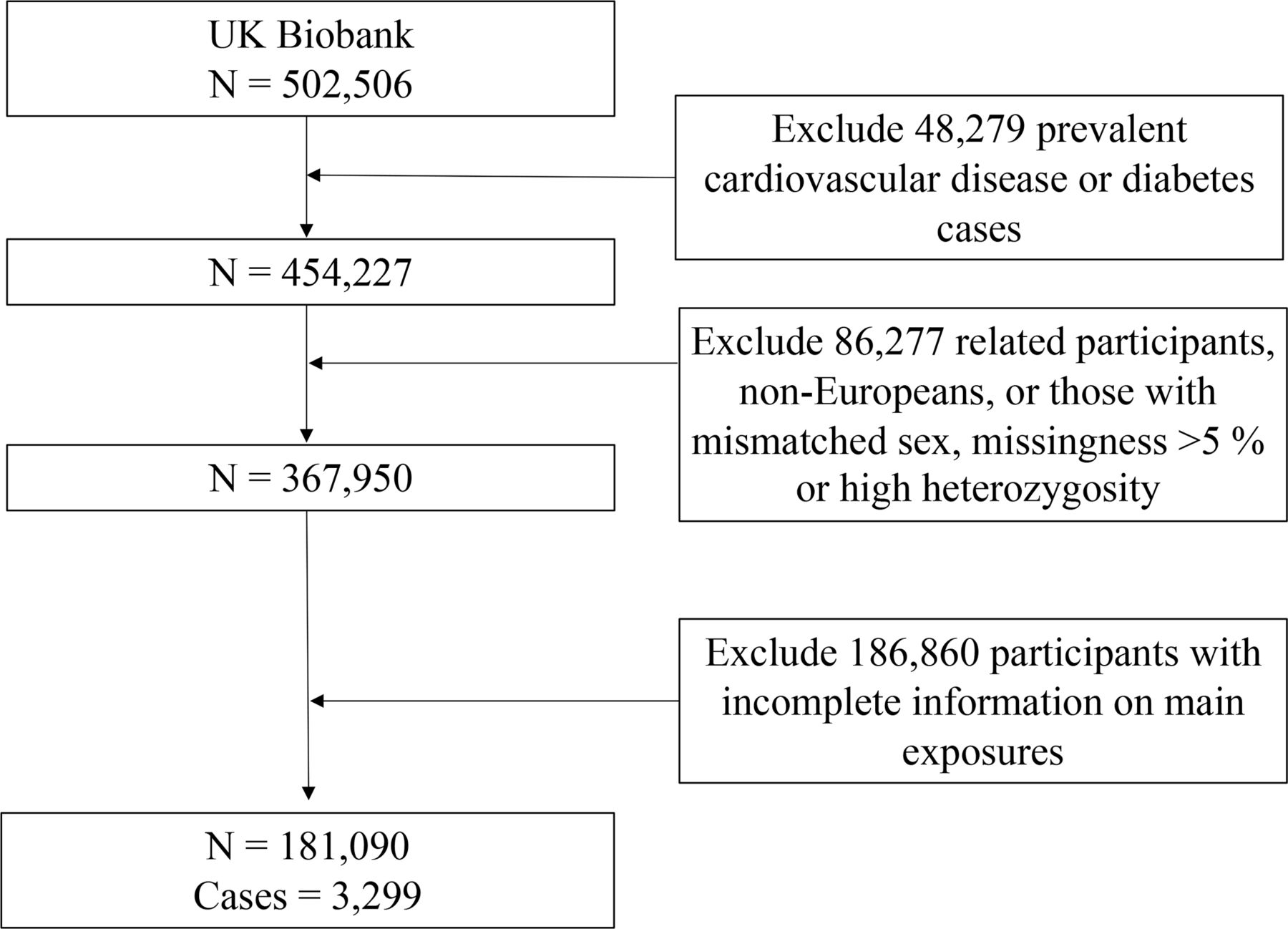
Details of the design of genotyping, quality control and imputation procedures are described previously.17 Briefly, the UK Biobank Axiom array was used to genotype around 450 000 participants, and UK BiLEVE array, which was closely related to the previous array, was used to genotype the other 5000 participants.17 In this study, we excluded the participants who were related, with excessive heterozygosity, high levels of missingness and mismatched sex, and those who were not Europeans (figure 1).

Flowchart for the selection of the study population from the UK Biobank.
We selected seven single-nucleotide polymorphisms (SNPs) associated with birth weight at genome-wide significance (p<5×10−8) in individuals of European ancestry in the Early Growth Genetics Consortium, which is present in online supplemental table 1).18 All the seven SNPs were available in the UK Biobank. The weighted GRS for each participant was defined using the formula weighted GRS=(β1×SNP1+β2×SNP2+β3×SNP3+…+βn×SNPn)×(N/sum of the β coefficients). Individual SNP was recoded based on the number of risk alleles (recoded as 0, 1 or 2). The beta-coefficient was the change in z score per birth weight lowering allele from linear regression according to the aforementioned consortium.18 The GRS ranged from 0.7 to 14.0, and a higher score indicated a higher genetic predisposition to low birth weight.
Supplemental material
Ascertainment of incident T2D cases
We used the first occurrence variables provided by UK Biobank to document T2D cases. The earliest occurrence dates have been extracted from multiple resources, including self-report data, hospital admission data, medical history data and death register data. The prevalent cases of all types of diabetes and major cardiovascular diseases including stroke, myocardial infarction, heart failure and ischemic heart disease were also identified by the first occurrence date variables and were excluded from the analysis. Detailed information on mapping the diagnosis and extracting the first occurrence date is available online (http://biobank.ndph.ox.ac.uk/showcase/showcase/docs/first_occurrences_outcomes.pdf).
Statistical analysis
Overall, the current analysis included data from 181 090 participants in the UK Biobank study after excluding the prevalent cases of T2D, or major cardiovascular disease, or those participants with incomplete information on main exposures, or those who were not Europeans, who were related, with excessive heterozygosity, high levels of missingness or mismatched sex (figure 1). Person-years were calculated from the date of recruitment to the date of diagnosis of T2D, death, loss to follow-up, or 1 March 2019, whichever occurred first. The missing values of covariates were imputed by sex-specific mean values for continuous variables or missing indicator approach for categorical variables (all covariates<8% missing). The differences between baseline characteristics across the quintiles of the circulating IGF-1 levels were examined using χ2 test for categorical variables and analysis of variance test for continuous variables among the individuals with birth weight of <2.5 and ≥2.5 kg.
We used multivariable Cox proportional hazards regression models to compute HRs and 95% CI for the association between circulating IGF-1 and risk of T2D using the lowest quintiles of IGF-1 as the reference group. Schoenfeld residuals method showed the proportional hazards assumption was not violated. To test for linear trends, we modelled the IGF-1 as a continuous variable by assigning the median values to all the participants in that category. We also analyzed the association between each 1-SD increment in the IGF-1 levels and the risk of T2D.
Three multivariate models were constructed. In model 1, we adjusted for the following potential confounders: age at recruitment (continuous, year), sex (men and women), overall health rating (excellent, good, fair, and poor), socioeconomic status (Townsend Deprivation Index, continuous), physical activity (continuous, metabolic equivalent of task-hours/week), maternal smoking (yes or no), birth weight (<2.5, ≥2.5 kg), smoking status (never, past, or current), alcohol consumption (never, monthly–weekly, or daily), sleep duration (≤6, 7–8, ≥9 hours/day), family history of diabetes (yes or no) and history of hypertension (yes or no). In model 2, we further adjusted for BMI (continuous, kg/m2). As evidence has suggested that the potential mechanisms between IGF-1 and risk of T2D may involve different aetiology pathways, including inflammation and lipid homeostasis,19 20 we further adjusted for the surrogate biomarkers of these pathways, namely, CRP, TC, TG and HDL-C in model 3. Genotyping batch and the first 10 principal components of ancestry were additionally adjusted in the models with GRS.
We then stratified the analysis by birth weight (<2.5, 2.5–<4.0 and ≥4.0 kg; <2.5 and ≥2.5 kg) overall and in men and women separately. We also examined the association between IGF-1 and T2D according to GRS of birth weight (Q1 vs Q2–Q5). The multiplicative interactions between IGF-1 and stratified factors (birth weight and GRS of birth weight) were tested using the likelihood ratio test by including an interaction term in the multivariable model.
We conducted a series of sensitivity analyses: (1) to overcome the potential underestimated observed associations using baseline measurements, we corrected for regression dilution ratio using the non-parametric method21 22; regression dilution ratio was calculated by the ratio of the mean range of the first repeat measurement to the mean range of the baseline measurement; (2) we additionally adjusted for baseline glycated hemoglobin (HbA1c) levels to minimize the influence of baseline impaired glucose tolerance; (3) we excluded the participants diagnosed with T2D within 2 years and other participants with less than 2 years of follow-up to minimize the potential effect of undiagnosed diabetes/pre-diabetes on circulating IGF-1 levels; (4) we repeated the same analyses among the participants in good or excellent health to minimize the potential confounding effect of subclinical diseases on the observed associations.
All analyses were performed using SAS V.9.4 and STATA statistical software V.14.0, and a two-sided p<0.05 was set as the threshold for statistical significance.
Results
During a mean follow-up of 9.9 (SD 1.4) years, we documented 3299 incident cases of T2D. Baseline characteristics of the participants across the quintiles of the circulating IGF-1 among the participants with birth weight of <2.5 and ≥2.5 kg are presented in table 1. In general, participants with lower levels of IGF-1 were older and more likely to be deprived, ever smokers, daily drinkers, have a higher BMI, experience short or longer sleep duration, report poor health rate, have a higher prevalence of pre-existing hypertension, maternal smoking exposure and a higher level of clinical biomarkers, including TC, TG and CRP.
Baseline characteristics according to quintiles of IGF-1 levels in the UK Biobank
Circulating IGF-1 levels and risk of T2D
Our data showed IGF-1 levels were inversely associated with the risk of T2D in a dose-dependent manner. With the adjustment for demographic, lifestyle risk factors and clinical biomarkers, compared with the lowest quintile, the HRs (95% CIs) of T2D for second quintile to the highest quintile were 0.85 (0.76–0.95), 0.84 (0.74–0.94), 0.72 (0.63–0.82) and 0.80 (0.70–0.92), respectively (p-trend<0.001). With each 1-SD increment in IGF-1 levels, the HR (95% CI) of T2D in the fully adjusted model was 0.93 (0.89–0.97) (table 2).
HRs (95% CI) of type 2 diabetes according to quintiles of IGF-1 levels
IGF-1 levels and risk of T2D according to the birth weight
We first analyzed the associations between circulating IGF-1 and the risk of T2D with the adjustment for the potential confounders according to the birth weight categorized as <2.5, 2.5–<4.0 and ≥4.0 kg. The association between IGF-1 and T2D was significantly modified by birth weight (p-interaction=0.02). As the risk estimates were similar in 2.5–<4.0 and ≥4.0 kg categories (online supplemental table 2), we combined them into a single category of birth weight of ≥2.5 kg. In the individuals with birth weight of ≥2.5 kg, IGF-1 levels were inversely associated with the risk of T2D in a dose-dependent manner (p-trend<0.001).
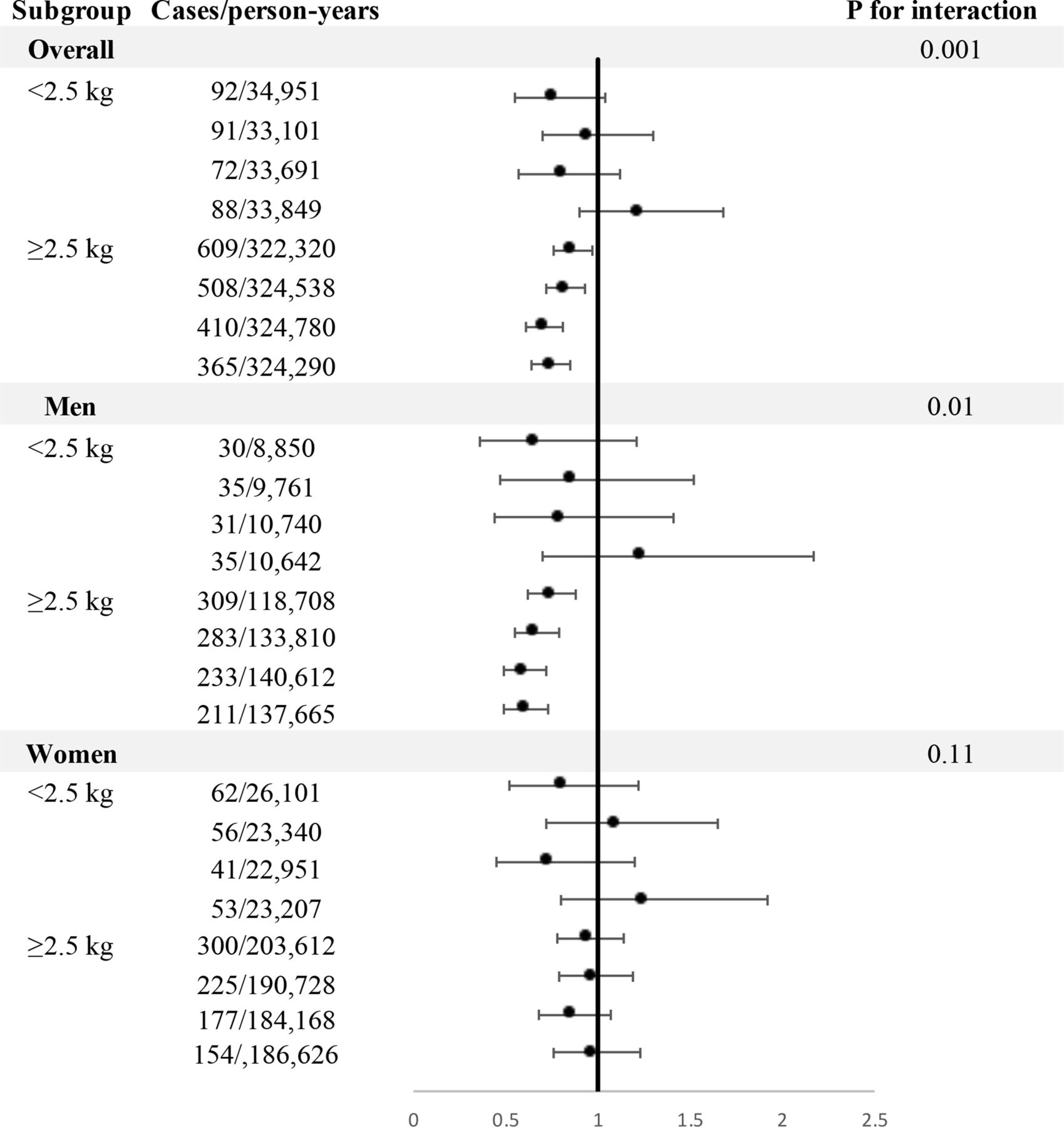
Compared with the lowest quintile, the HRs of T2D were 0.86 (95% CI 0.76 to 0.97) for the second quintile, 0.82 (95% CI 0.72 to 0.93) for the third quintile, 0.71 (95% 0.61 to 0.81) for the fourth quintile, and 0.74 (95% CI 0.64 to 0.85) for the highest quintile. Each 1-SD increment in circulating IGF-1 was statistically associated with a 9% lower risk of T2D. In contrast, IGF-1 was not associated with the risk of T2D in individuals with birth weight of <2.5 kg (p-interaction=0.001). The interaction between IGF-1 and birth weight on T2D was observed in men (p-interaction=0.01) but not in women (p-interaction=0.11). In men, an inverse association between IGF-1 and risk of T2D was observed among the participants with birth weight of ≥2.5 kg (p-trend<0.001), whereas the association was attenuated to be not significant among those whose birth weight was <2.5 kg (p-trend=0.61) (figure 2).

{kind=link}
{kind=link}
Associations between insulin-like growth factor-1 and risk of type 2 diabetes stratified by birth weight. HRs were adjusted for age, sex, body mass index, overall health rating, Townsend Deprivation Index, smoking status, alcohol consumption, sleep duration, physical activity, maternal smoking, menopause status and hormone-replacement therapy (women only), family history of diabetes, history of hypertension, circulating concentrations of C reactive protein, total cholesterol, triglycerides and high-density lipoprotein cholesterol.
IGF-1 levels and risk of T2D according to the GRS of birth weight
We did not observe a significant interaction between IGF-1 levels and the GRS of birth weight in relation to the risk of T2D (p-interaction=0.47). Compared the highest with the lowest quintiles, the multivariable adjusted HRs for T2D was 0.82 (95% CI 0.61 to 1.09) for those at the lowest quintile of the GRS of birth weight and 0.81 (95% CI 0.70 to 0.95) for those at the highest 80% of the GRS (table 3).
HRs (95% CI) of type 2 diabetes according to quintiles of IGF-1 levels stratified by GRS of birth weight
Sensitivity analyses
The results were generally robust in the sensitivity analyses correcting for regression dilution ratio and adjusting for the additionally adjusted for baseline HbA1c levels (data not shown). The results were essentially unchanged after excluding the T2D cases diagnosed within 2 years and non-cases with less than 2 years of follow-up (online supplemental table 3) or among those who reported good or excellent overall health rating (online supplemental table 4).
Discussion
To our best knowledge, this is the first study to provide epidemiological evidence that the relationship between circulating IGF-1 and risk of T2D was significantly modified by birth weight. Our data showed an inverse association between IGF-1 and risk of T2D in individuals with birth weight of ≥2.5 kg, but not in those with birth weight of <2.5 kg. Such effect modification was not observed for the GRS of birth weight.
Our findings of the inverse associations between circulating IGF-1 and T2D are consistent with the results from several epidemiological studies.23–25 Null associations were also observed between IGF-1 and T2D.26 27 The discrepant observations might be partly due to the small sample size (less than 800 cases) or failure in excluding baseline T2D prevalent cases. Moreover, a recent Mendelian randomization study showed that the genetic predisposition to elevated IGF-1 levels during adulthood was associated with a higher risk of T2D, suggesting a potentially causal relation.13 However, in this study, some SNPs used as instrumental variables were also related with other traits that may confound the relationship between IGF-1 and risk of T2D, such as hypertension, vascular or heart disease, birth weight of the first child, body fat mass, and height. Hence, caution is needed when interpreting the results of the Mendelian randomization study.
The significant interaction between birth weight and circulating IGF-1 on T2D is in line with our previous findings that birth weight may modify the adulthood risk factors on cardiometabolic disease risk.6 28 Of note, data on the relations between IGF-1 and T2D risk from the previous observational studies are highly conflicting, with findings of inverse,23–25 U-shaped,29 or null associations.26 27 30 31 Such heterogeneous results suggest the potential effect modifications may exist. Our results indicate that birth weight is likely to be an effect modifier on the relation between IGF-1 and T2D.
An inverse association between IGF-1 and T2D was only observed among the participants with birth weight of ≥2.5 kg, while the association between IGF-1 and T2D was attenuated toward null in those with birth weight of <2.5 kg. The putative effect modification by birth weight on the relationship between IGF-1 and T2D might be explained by several potential biological mechanisms. Low birth weight is a widely accepted indicator for prenatal malnutrition and intrauterine growth restriction. Evidence has shown that intrauterine growth restriction may lead to developmental programming of lifecourse IGFs concentrations. It was found that intrauterine growth restriction was associated not only with lower levels of cord IGF-132 but also with decreased levels of IGF-1 in infants,33 preadolescent children without significant ‘catch-up’ growth34 and adults.12 Experimental studies indicate that intrauterine growth restriction can result in persistent reduction of serum IGF-1 through modifying epigenetic characteristics, especially the histone code of the hepatic IGF-1 gene,35 perturbing developmental epigenetics around distal growth hormone response elements on hepatic IGF-1 gene,36 and disturbing the formation of nucleosome-depleted region at the right potion on the IGF-1 gene.37 Moreover, prenatal malnutrition had long-term effects on glucose–insulin metabolism through multiple pathways involving IGFs, including epigenetic modifications, permanent changes in organs’ structure during a sensitive period of development and persistent effects on cellular aging regulation.11 38 Hence, we assumed that the effects of elevated IGF-1 levels could be nullified by these aforementioned alterations caused by prenatal malnutrition and intrauterine growth restriction.
In addition, our data showed the association between IGF-1 and T2D was not modified by genetic predisposition to birth weight. As the GRS used in our study only explained 2% of the variance in birth weight,39 the non-significant interaction may be due to the genetic variations that only account for a small proportion of the variance in birth weight. Further studies are warranted to corroborate our findings on the synergistic effect of GRS of birth weight and IGF-1 levels in adulthood on the risk of T2D.
A body of evidence from animal and human studies suggests that the early life development windows are critical periods when environmental exposure is more harmful to health outcomes throughout the life course.40 In normal condition, IGF-1 can act independently or coordinate with insulin to maintain the glucose homeostasis.41 However, our data showed the potential beneficial effect of elevated IGF-1 in adulthood on T2D might be influenced by the impaired fetal growth. The findings in this present study suggest that improvement of prenatal/postnatal exposures plays a critical role in the prevention and treatment of T2D through the pathway involving IGF-1.
The major strengths of this study include the prospective study design, large sample size, and comprehensively collected information on the lifestyle factors. Moreover, the value of regression dilution ratio derived from the baseline and repeated measurements of IGF-1 indicated that the baseline IGF-1 levels could provide a precise estimate of long-term IGF-1 levels. Our study also has several potential limitations. First, information regarding IGF-binding proteins and IGF-2 was not available in the UK Biobank; hence, we were unable to adjust for these factors as confounders or mediators in the model. We were unable to investigate the interaction between free IGF-1 and birth weight on T2D as well. Second, covariates collected at baseline could have changed over time; however, given the prospective study design, the subsequent misclassification would be non-differential, which would most likely to attenuate the true associations. Third, although data on birth weight was self-reported in our study, a previous study has validated the variable and demonstrated that the self-reported birth weight in the UK Biobank was a reliable measurement.16 Furthermore, as UK Biobank did not collect the information on birth terms, we were unable to further categorize the birth weight on the basis of preterm or full-term birth. Fourth, due to the observational study design, our data could not be used for inference of causality. Finally, evidence has shown that the ethnicity may affect the circulating IGF-1 levels42; hence, studies in other ethnic populations are needed.
Our data indicate that the relation between adulthood IGF-1 and T2D is significantly modified by birth weight. Our findings highlight the importance of early-life risk factors in the development of the lifecourse prevention strategies targeting on IGF-1 and T2D.
Acknowledgments
We are grateful to all the participants of UK Biobank and all the people involved in building the UK Biobank study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Press_Release
Footnotes
Contributors LQ designed the study and had primary responsibility for the final content; TG and MW performed the statistical analysis; TG wrote the paper with critical input from all authors. All authors approved the manuscript.
Funding The study was supported by grants from the National Heart, Lung, and Blood Institute (HL071981, HL034594, and HL126024) and the National Institute of Diabetes and Digestive and Kidney Diseases (DK115679, DK091718, DK100383, and DK078616).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The UK Biobank study was approved by the National Information Governance Board for Health and Social Care in England and Wales, and the Community Health Index Advisory Group in Scotland and the North West Multicenter Research Ethics Committee. All participants gave written informed consent. This present analysis was approved by the Tulane University (New Orleans, Louisiana) Institutional Review Board (study number: 2018–1872).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. This research has been conducted using the UK Biobank Resource under Application Number 29256.The UK Biobank data are available on application to the UK Biobank (www.ukbiobank.ac.uk/).
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.