Article Text

Abstract
Introduction Insulin icodec is a novel, long-acting insulin analog designed to cover basal insulin requirements with once-weekly subcutaneous administration. Here we describe the molecular engineering and the biological and pharmacological properties of insulin icodec.
Research design and methods A number of in vitro assays measuring receptor binding, intracellular signaling as well as cellular metabolic and mitogenic responses were used to characterize the biological properties of insulin icodec. To evaluate the pharmacological properties of insulin icodec in individuals with type 2 diabetes, a randomized, double-blind, double-dummy, active-controlled, multiple-dose, dose escalation trial was conducted.
Results The long half-life of insulin icodec was achieved by introducing modifications to the insulin molecule aiming to obtain a safe, albumin-bound circulating depot of insulin icodec, providing protracted insulin action and clearance. Addition of a C20 fatty diacid-containing side chain imparts strong, reversible albumin binding, while three amino acid substitutions (A14E, B16H and B25H) provide molecular stability and contribute to attenuating insulin receptor (IR) binding and clearance, further prolonging the half-life. In vitro cell-based studies showed that insulin icodec activates the same dose-dependent IR-mediated signaling and metabolic responses as native human insulin (HI). The affinity of insulin icodec for the insulin-like growth factor-1 receptor was proportionately lower than its binding to the IR, and the in vitro mitogenic effect of insulin icodec in various human cells was low relative to HI. The clinical pharmacology trial in people with type 2 diabetes showed that insulin icodec was well tolerated and has pharmacokinetic/pharmacodynamic properties that are suited for once-weekly dosing, with a mean half-life of 196 hours and close to even distribution of glucose-lowering effect over the entire dosing interval of 1 week.
Conclusions The molecular modifications introduced into insulin icodec provide a novel basal insulin with biological and pharmacokinetic/pharmacodynamic properties suitable for once-weekly dosing.
Trial registration number NCT02964104.
- diabetes mellitus
- experimental
- insulin
- pharmacokinetics
- pharmacology
Data availability statement
For the non-clinical studies, data associated with this work are present in the paper. Data and reagents may be made available, on reasonable request, from Novo Nordisk A/S under a materials transfer agreement. For the clinical trial, individual participant data will be shared in data sets in a deidentified/anonymized format. Datasets from Novo Nordisk sponsored clinical research completed after 2001 for product indications approved in both the EU and USA will be shared. Trial protocol and redacted Clinical Trial Report will be available according to Novo Nordisk data sharing commitments. The data will be available permanently after research completion and approval of product and product use in both EU and USA. Data will be shared with bona fide researchers submitting a research proposal requesting access to data. Data will be shared for analyses as approved by the Independent Review Board (IRB) according to the IRB Charter (see novonordisk-trials.com). Data will be available through an access request proposal form, and the access criteria can be found at novonordisk-trials.com. The data will be made available on a specialized SAS data platform.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Basal insulin therapy, currently administered either once or twice daily, is a very well-established option for the treatment of diabetes.
Acylation has been proven to be an applicable technology to engineer the once-daily basal insulin analogs, insulin detemir and insulin degludec, as well as longer acting insulin analogs such as oral insulin 338.
What are the new findings?
Insulin icodec, a new acylated basal insulin analog, has been engineered with optimized modifications to give a long half-life suitable for once-weekly insulin administration.
Strong albumin binding combined with reduced insulin receptor affinity ensures slow clearance and the formation of an essentially inactive albumin-bound depot, providing slow and continuous insulin action.
Insulin icodec retains the same biological properties as natural human insulin, with no increase in insulin-like growth factor-1 receptor binding or mitogenicity.
In a clinical pharmacology trial in people with type 2 diabetes, insulin icodec was well tolerated and had a half-life of 196 hours with mean glucose-lowering effect being close to evenly distributed over the entire week.
How might these results change the focus of research or clinical practice?
Insulin icodec may offer a convenient, safe and efficacious once-weekly basal insulin option that could eventually be widely used in clinical practice for the treatment of diabetes.
Introduction
Despite the lifesaving effects and unsurpassed glucose-lowering capability of insulin, the overall glycemic control observed in people with type 1 diabetes and type 2 diabetes does not meet recommended targets.1–4 The challenge in achieving good glycemic control with insulin therapy can be attributed to the complexity associated with matching the dose and timing of daily insulin injections to the actual physiological requirement.5 For people with type 1 diabetes, there are currently no options other than insulin therapy administered as multiple daily insulin injections or via pumps. For people with type 2 diabetes, there are a number of alternatives, including oral antidiabetic medicines and injectables such as glucagon-like peptide-1 receptor agonists (GLP-1RAs). Although glucose regulation often becomes inadequate with these options as the disease progresses, there is some degree of ‘clinical inertia’ due to the complexity and fear of insulin therapy, both from the perspectives of healthcare providers and those with diabetes.6 7
Delaying the start of insulin therapy in type 2 diabetes contributes to overall poor glycemic control.8 A very long-acting basal insulin that can safely and effectively provide consistent glucose lowering with only one injection per week should reduce the treatment burden, resulting in more people in need of better glucose regulation to actually initiate insulin therapy. This would expectedly lead to better adherence to therapy and an overall improvement in glycemic control.9 10 For people with type 1 diabetes, a once-weekly basal insulin could be seen as an attractive alternative to daily basal insulin injections, particularly if adherence is an issue.
For a once-weekly basal insulin to be clinically relevant, it must be as effective as conventional once-daily insulin treatment and above all should not lead to increased risk of hypoglycemia when administering 7 days’ worth of insulin in one injection. Recent advances in molecular engineering of insulin analogs have achieved very long plasma elimination half-lives in humans as described for the acylated oral insulin 338.11 12 Here, we have further engineered this concept for subcutaneous delivery of a once-weekly basal insulin analog: insulin icodec. Introducing strong, reversible binding to albumin, together with reduced insulin receptor (IR) affinity to slow down clearance, ensures that insulin icodec forms a circulating albumin-bound depot, which is essentially inactive, from which insulin icodec can slowly and consistently activate IRs, resulting in a long half-life suitable for once-weekly administration. A clinical pharmacology trial demonstrates that insulin icodec has a half-life of 196 hours and a glucose-lowering effect that is close to evenly distributed throughout the week, thus making it indeed suitable for once-weekly dosing.
Research design and methods
Production of insulin icodec
The insulin icodec precursor was expressed in Saccharomyces cerevisiae as a single-chain with a mini-C-peptide linking the B-chain (B1–B29) to the A-chain (A1–A21).13 After purification, the single-chain precursor was enzymatically converted into two-chain insulin A14E, B16H, B25H, desB30.14 Insulin icodec was chemically modified by coupling the N-hydroxysuccinimide-activated side chain to the epsilon amino group of B29 lysine.15
Albumin binding studies
The binding of [125I]-insulin icodec to human serum albumin (HSA) was determined in a binding assay as previously described for insulin detemir.16 Briefly, [125I]-insulin icodec was mixed with increasing amounts of HSA conjugated to agarose. Free radioactivity was measured, and the ratio of bound/free radioactivity was plotted against the HSA concentration to obtain the affinity (slope) of insulin icodec for HSA. Insulin detemir was used as a comparator.
Receptor binding studies
IR and insulin-like growth factor-1 receptor (IGF-1R) binding studies were performed essentially as described previously.17 Briefly, receptor affinities were determined by competition scintillation proximity assays using either solubilized IRs or IGF-1Rs in the absence or presence of HSA. Concentration-response relations from the binding assays were analyzed using a four-parameter logistic model18 assuming common slope and basal and maximum response.
Intracellular signaling studies
To study intracellular signaling, Chinese Hamster Ovary (CHO) cells overexpressing human insulin receptors (hIRs) were used.19 Phosphorylation of the IR, extracellular-regulated kinase (ERK) and protein kinase B (AKT) were measured after stimulation with increasing concentrations of HI or insulin icodec. The experiments were performed in the presence of 0.1% HSA.
Cellular metabolic effects
In primary rat adipocytes isolated from epididymal fat pads, the effect of insulin icodec on lipogenesis was determined by measuring the incorporation of [3H]-labeled glucose into fat essentially as described previously.20 Glycogen accumulation was measured in rat hepatocytes isolated using a two-step collagenase perfusion technique.21 Cells were incubated with varying concentrations of HI or insulin icodec in the presence or absence of HSA. Cellular glycogen content was determined by measuring glucose following digestion by amyloglucosidase. Glycogen synthesis was determined in L6-hIR cells (rat skeletal myoblast L6 cell line stably transfected with the A isoform of the hIR), as well as in MCF-7 cells (human mammary adenocarcinoma cell line), as previously described.22 The effect of insulin icodec relative to HI was determined in these two cell lines by measuring glycogen synthesis as incorporation of [14C]-labeled glucose into glycogen.
Cellular mitogenic responses
The mitogenic response to insulin icodec was examined in L6-hIR and MCF-7 cells and in human mammary epithelial cells (HMECs) and human colorectal adenocarcinoma cells (COLO-205) by measuring the cellular incorporation of [6-3H]-thymidine as indication of DNA synthesis and thus cell proliferation. Mitogenicity in HMEC and MCF-7 was performed as previously described.23 24 Mitogenicity in COLO-205 cells was measured as described for MCF-724 with minor modifications: 1.2×105 COLO-205 cells were seeded per well directly in assay medium for 24 hours before stimulation. Mitogenicity in L6hIR cells was measured as for COLO-205 except that 4×104 cells were seeded per well, Dulbecco’s Modified Eagle Medium (DMEM) with low glucose was used and cells were stimulated for 6–8 hours before addition of [6-3H]-thymidine for additional 15–17 hours.
Pharmacokinetic profiles in dogs
Beagle dogs weighing 9–18 kg were not offered food for at least 12 hours prior to dosing subcutaneously in the neck with the different insulin analogs. Blood samples were drawn regularly for 7–14 days. Time points for blood sampling were prespecified for each analog depending on the expected pharmacokinetic profile. Plasma samples were analyzed for the respective insulin analog by luminescent oxygen channeling immunoassay (AlphaLISA) using antibodies specific for each analog tested.25 Plasma concentration–time profiles were analyzed by non-compartmental pharmacokinetics analysis using WinNonlin Professional. Calculations were performed using concentration–time values from each animal.
Clinical pharmacology trial in individuals with type 2 diabetes
To evaluate the pharmacological properties of insulin icodec in individuals with type 2 diabetes, a randomized, double-blind, double-dummy, active-controlled, multiple-dose, dose escalation trial was conducted. Eligible participants had type 2 diabetes, were treated with any insulin (total daily dose 0.3–1.0 U/kg), aged 18–64 years, with glycosylated hemoglobin (HbA1c) ≤9.0% (75 mmol/mol) and were not using oral antidiabetic drugs or GLP-1RAs (subjects on insulin therapy in combination with metformin were allowed in cohort 3, but metformin was washed out for ≥3 weeks prior to first trial product administration in these subjects).
Within each of three subsequent cohorts, participants were randomized to once-weekly insulin icodec (cohort 1: 12 nmol/kg; cohort 2: 20 nmol/kg; cohort 3: 24 nmol/kg) plus once-daily placebo or once-daily insulin degludec (0.4 U/kg; corresponding to 16.8 nmol/kg/week) plus once-weekly placebo for 5 weeks (all by subcutaneous injection in the thigh at ~20:00 hours using a NovoPen4 pen device; Novo Nordisk A/S, Bagsværd, Denmark) (online supplemental figure 1). Data on insulin degludec are not included, but pharmacodynamic data have been presented elsewhere.26 The decision to proceed to the next cohort was made by an internal trial safety group after review of blinded interim safety, pharmacokinetic and pharmacodynamic data. Total serum insulin icodec concentration (both the free and albumin bound) was measured using a specific assay at prespecified time points from predose until 36 days after the last weekly insulin icodec dose (online supplemental table 1). Steady-state pharmacodynamic properties were assessed during two 24-hour automated euglycemic glucose clamps (ClampArt, Profil, Neuss, Germany; target of 5.5 mmol/L) at steady state on day 2 (24–48 hours) and day 7 (144–168 hours) after the last weekly insulin icodec dose in week 5 (ie, days 30 and 35 of the treatment period; online supplemental figure 1). Safety assessments included adverse events, hypoglycemic episodes, injection site reactions, vital signs, physical examination, clinical laboratory assessments and ECG.
Supplemental material
Pharmacokinetic dose proportionality was evaluated by linear regression of log(AUC0-168h) on log(dose), where AUC0-168h was derived after the last insulin icodec dose and a slope of 1 corresponds to dose proportionality (the power model). Pharmacokinetic trough values to steady state and full-week glucose-lowering effect profiles in week 5 were predicted for each individual using a pharmacokinetic–pharmacodynamic model. Model parameters were estimated for each individual based on insulin icodec pharmacokinetic data from the first dose until day 65 and 24-hour glucose clamp data in week 5 (days 30 and 35).
Results
Molecular design of insulin icodec
Engineering of an insulin molecule suitable for once-weekly administration requires not only a very long half-life, but just as importantly a mechanism to ensure that the glucose-lowering effect is close to evenly distributed throughout the week. To achieve this, insulin icodec was designed to introduce four key features: high affinity for albumin, improved stability, low IR binding affinity and high solubility. Very strong, reversible albumin binding was achieved by introducing a 20-carbon atom long icosane fatty diacid (icosanedioic acid) at the C-terminal of the B-chain of the HI amino acid sequence (figure 1A). The icosanedioic acid is connected via a hydrophilic linker (γ-glutamic acid and a small PEG-like linker) to the lysine in position B29. The fatty diacid moiety of insulin icodec binds strongly to fatty acid binding sites on albumin, and the affinity of insulin icodec for albumin was estimated to be 9.5±0.6-fold greater (mean±SEM, n=3) than that of insulin detemir, which was used as a reference (online supplemental figure 2). However, the C20 fatty diacid was not sufficient to prolong the half-life to the extent needed for once-weekly dosing. Thus, three amino acid substitutions, TyrA14Glu (A14E), TyrB16His (B16H) and PheB25His (B25H), were introduced in order to lower the IR binding and subsequently IR mediated clearance to confer a longer half-life. Each individual amino acid substitution contributes by lowering the IR affinity (A14E: to 80%, B16H: to 22%, B25H: to 30% relative to HI) such that the IR affinity of the molecule without the chemical modification is 5.5% relative to HI. The high albumin binding affinity of the fatty diacid limits the number of insulin icodec molecules available to bind to the IR further reducing the relative affinity from 5.5% to 0.03% in the presence of 1.5% HSA. Furthermore, the amino acid modifications confer molecular stability by minimizing enzymatic degradation.11 15 Finally, the threonine in position B30 of HI has been deleted; however, the desB30 has no influence on the IR affinity. Altogether, these molecular changes have been demonstrated to impart a long half-life as seen by the pharmacokinetic profile of insulin icodec in dogs following a single subcutaneous injection (online supplemental figure 2).
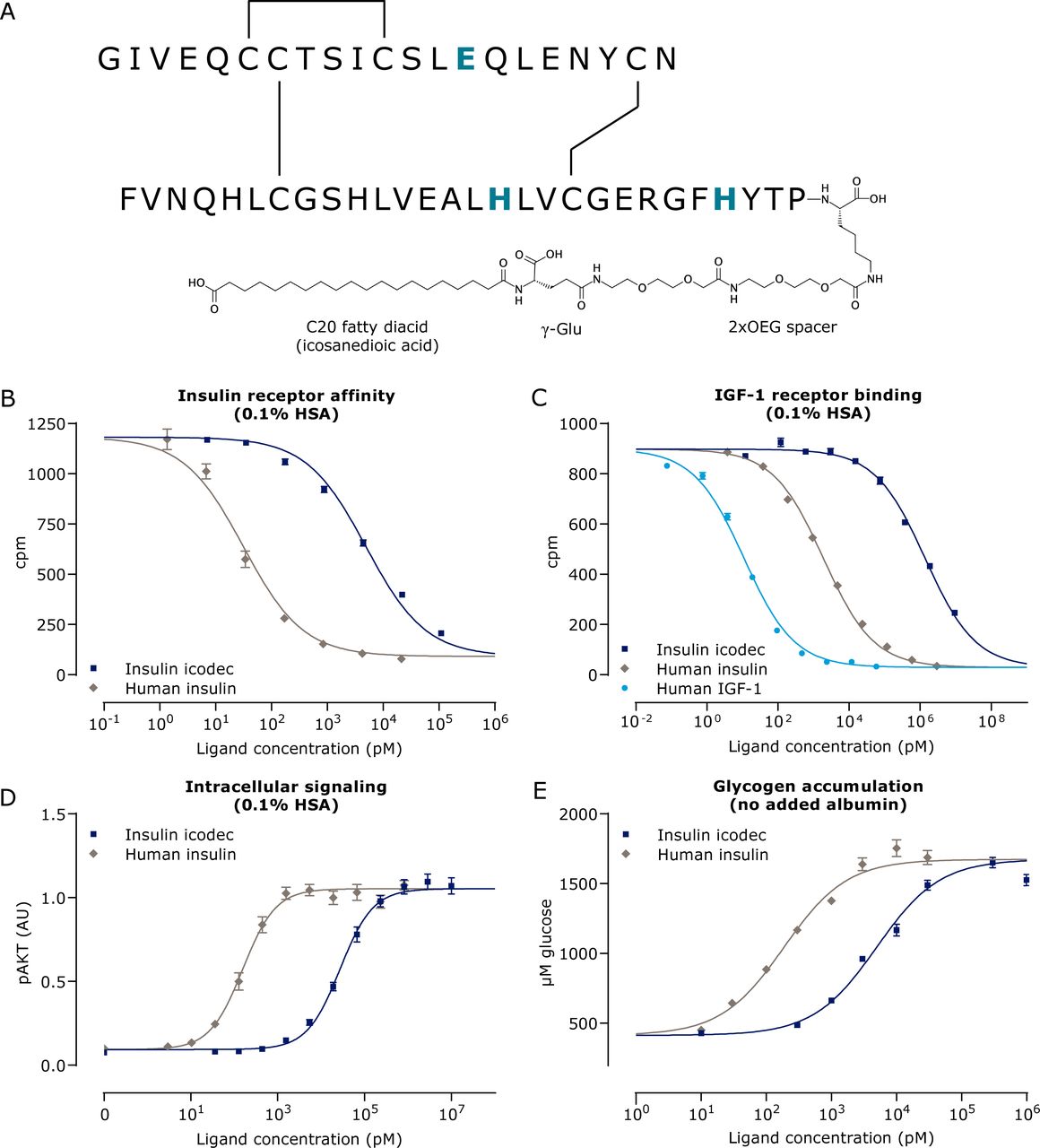
Schematic depiction and biological properties of insulin icodec. (A) Insulin icodec structure showing changes to the human insulin amino acid sequence and chemical modification attached to the lysine in position B29 of insulin. Insulin substitutions relative to human insulin are shown in blue and bold (TyrA14Glu, TyrB16His and PheB25His). (B) Displacement curves for binding to solubilized hIR (n=4). (C) Displacement curves for binding to solubilized hIGF-1R (n=4). (D) Phosphorylation of the IR downstream signaling molecule AKT in CHO-hIR cells (n=3). (E) Accumulation of glycogen in rat hepatocytes (n=7). Error bars show SEM. Lines show the non-linear fit. AKT, protein kinase B; AU, arbitrary units; CHO-hIR, Chinese Hamster Ovary cells overexpressing hIR; γ-Glu, γ-glutamic acid; hIGF-1R, human insulin-like growth factor-1 receptor; hIR, human insulin receptor; HSA, human serum albumin; IR, insulin receptor; OEG, oligoethylene glycol.
Moreover, A14E in combination with B25H was previously reported to improve solubility,11 also an important feature of insulin icodec, allowing for a 4.2 mM formulation, that is, seven times higher than a standard U100 insulin formulation. This ensures that the once-weekly insulin icodec dose volume can be low and similar to once-daily basal insulin dosing volumes.
Biological characterization of insulin icodec
A key consideration when developing insulin analogs is to ensure that the modifications to achieve the desired pharmacokinetic profile do not unfavorably alter the biological responses. Several in vitro studies were therefore conducted to demonstrate that insulin icodec retains the biological properties of native HI. Similar assays have been previously used to assess the biological characteristics and in vitro safety profiles of various long-acting basal insulin analogs (insulin glargine, detemir and degludec).23 27 28 From the displacement curves for binding to the solubilized hIR, it can be seen that insulin icodec fully displaces the HI tracer to the same extent as HI (figure 1B). However, the relative binding in the presence of 1.5% HSA is approximately 0.03% that of HI, indicating low affinity (table 1). The difference in binding affinity obtained in the presence or absence of HSA reflects insulin icodec’s ability to bind albumin. Binding to the structurally related IGF-1R is correspondingly low (figure 1C and table 1). Insulin icodec is a full agonist and was shown to activate IRs and stimulate downstream signaling molecules AKT (figure 1D) and ERK in a similar manner as HI (table 1).
Summary of the receptor binding and signaling properties of insulin icodec
Insulin icodec was shown to have the same cellular metabolic profile as HI, stimulating glucose uptake and lipogenesis in rat adipocytes and glycogen accumulation in rat hepatocytes (figure 1E and table 2). Insulin icodec also stimulated glycogen synthesis in L6-hIR and MCF-7 cells (table 2).
Overview of in vitro assays demonstrating that insulin icodec has retained the same biological effects as human insulin
Since native HI has cell proliferative effects in addition to its metabolic actions, it is important to determine both the metabolic and mitogenic effects of new insulin analogs and the relative balance between these two effects. The mitogenic response of insulin icodec was examined in various cell types that express IR and IGF-1R to differing degrees, including L6-hIR, MCF-7, COLO-205 and HMEC cells. As can be seen in table 2, the mitogenic potencies (all expressed as a percentage relative to HI) are within the same range as the metabolic potencies. It should be noted that it was not possible to conduct all metabolic and mitogenic cellular assays under the same conditions, particularly the same albumin concentration, as different cell types have differing sensitivities to albumin. Taking this into consideration, the overall data indicate that insulin icodec has retained a balanced mitogenic-to-metabolic potency ratio.
Clinical pharmacological properties of insulin icodec
In the clinical pharmacology trial, 38 individuals were randomized to receive insulin icodec 12 nmol/kg (n=13), 20 nmol/kg (n=13) or 24 nmol/kg (n=12). The 38 individuals (33 men) had a mean±SD age of 57.8±4.3 years, body weight of 93.9±13.3 kg, body mass index of 29.9±2.8 kg/m2, HbA1c of 7.4%±0.6% and duration of diabetes of 14.4±6.7 years with comparable treatment groups (online supplemental table 2). In cohort 3, seven participants (58.3%) used metformin at screening.
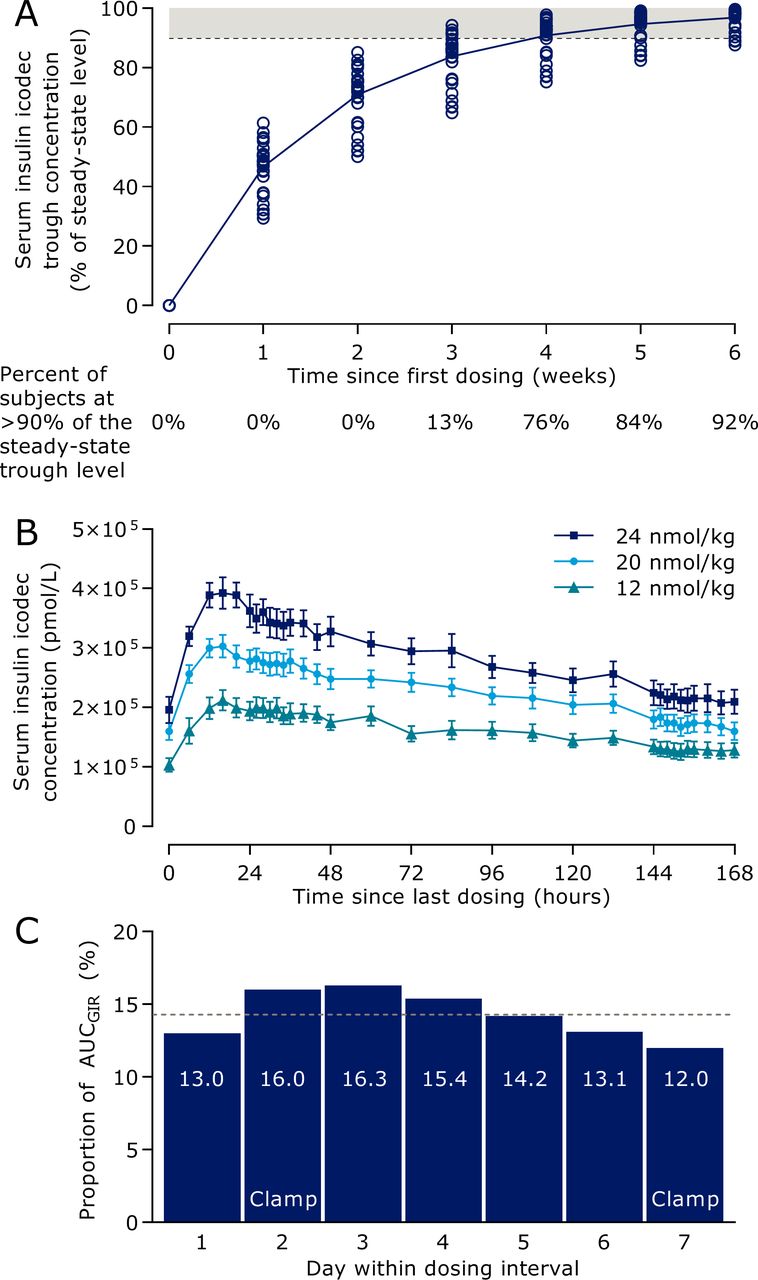
Model-predicted insulin icodec trough concentrations during initiation of once-weekly dosing (figure 2A) indicated that in the majority of individuals, it takes 3–4 weeks of once-weekly insulin icodec dosing to achieve clinical steady state, defined as a trough concentration above 90% of the steady-state trough level.29 The mean total serum insulin icodec profiles after multiple dosing showed characteristics suited for once-weekly administration (figure 2B). Geometric mean half-life was 196 hours and median time to maximum concentration was 16 hours, with no systematic differences between dose levels. Results supported dose-proportionality for AUC0-168h as the slope of log(AUC0-168h) versus log(dose) was not statistically significantly different from 1 (0.83 (0.56; 1.10)95% CI). Model-predicted pharmacodynamic data of a 1 week dosing interval at steady state indicated rather consistent glucose-lowering effect of insulin icodec throughout the week at clinically relevant doses (figure 2C). The distribution of glucose-lowering effect of insulin icodec over a week at steady state was determined as the daily proportion of the total weekly model-predicted glucose-lowering effect. The dotted line in figure 2C represents equal distribution across 7 days corresponding to a daily proportion of 14.3%, and the range over the week was from 12.0% to 16.3% per day, hence only deviating −2.3% points to +2.0% points from complete even distribution.
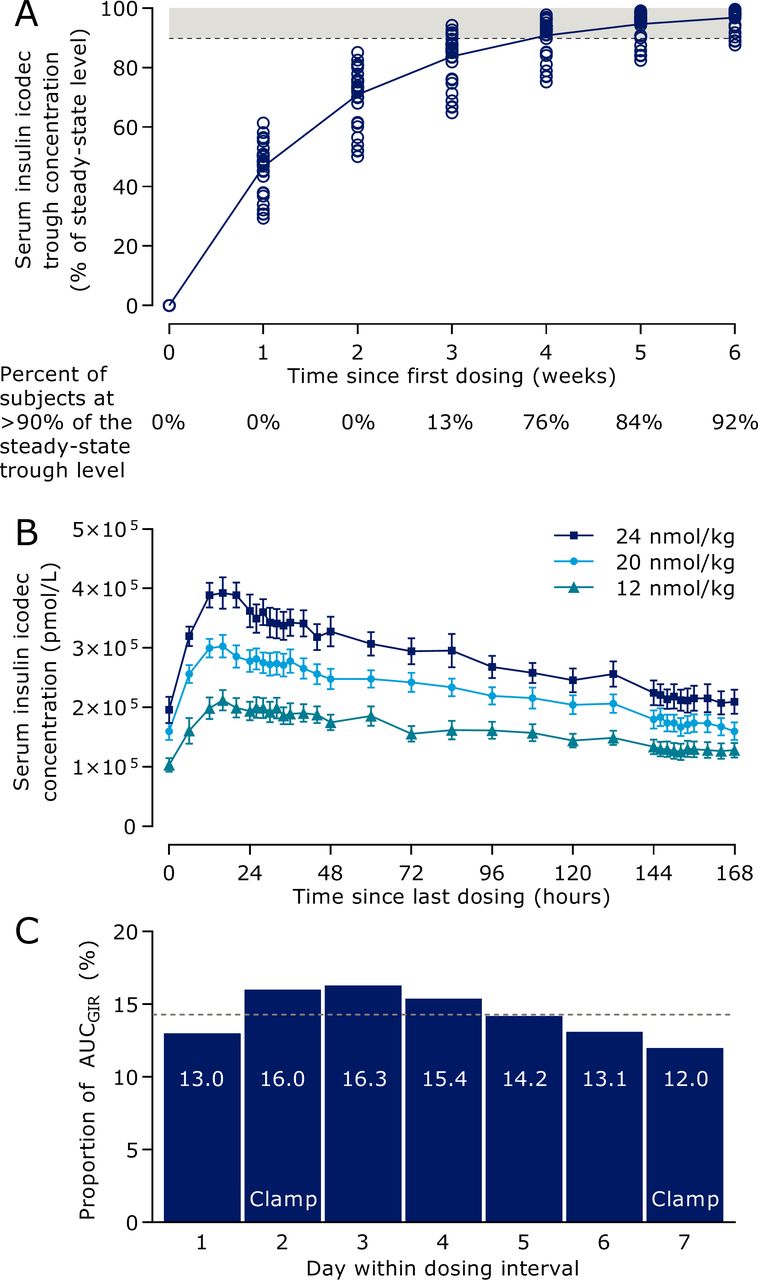
Pharmacokinetic and pharmacodynamic properties of insulin icodec in individuals with type 2 diabetes. (A) Model-predicted serum insulin icodec trough concentration during initiation of once-weekly dosing. The dashed line indicates the threshold for clinical steady state of serum insulin icodec and the shaded area indicates serum insulin icodec concentrations considered as clinical steady state. Circles indicate individual values (n=38). (B) Mean observed total serum insulin icodec concentration (the vast majority being albumin-bound) during week 5 of once-weekly dosing. Error bars show standard error of the mean (n=38). Insulin icodec once-weekly doses correspond to 0.29, 0.48 and 0.57 U/kg/day anticipating equipotency to once-daily basal insulin. (C) Model-predicted distribution of glucose-lowering effect of insulin icodec within a dosing interval at steady state. Dotted line represents equal distribution across 7 days. All three dose levels are combined. Data are arithmetic mean (n=32). AUC, area under the curve; GIR, glucose infusion rate.
Insulin icodec was well tolerated with no increase in adverse event incidence with increasing insulin icodec doses (12 nmol/kg: 33 events in 13 individuals; 20 nmol/kg: 19 events in 9 individuals; 24 nmol/kg: 23 events in 9 individuals). No serious or severe adverse events, severe hypoglycemic episodes, injection site reactions or clinically significant findings in vital signs, physical examination, clinical laboratory assessments or ECG were observed.
Discussion
Bearing in mind that optimal insulin therapy requires very precise dosing in terms of both the actual dose and timing, it may be conceptually difficult to appreciate that it would be possible to safely administer an entire week’s worth of basal insulin in a single injection without risking hypoglycemia. The design of the insulin icodec molecule addresses this concern by introducing modifications to the insulin molecule that not only extend insulin action to cover an entire week, but most importantly slow down the effect to a ‘trickle’ by ensuring that the given dose enters an essentially inactive reservoir (albumin bound) from which a slow and steady glucose-lowering effect is achieved. The application of fatty acid acylation technology combined with amino acid substitutions to reduce IR affinity in order to obtain prolongation of plasma half-lives by reducing the rate of receptor mediated clearance of insulin molecules has recently been described for oral basal insulin analogs.11 Insulin icodec was engineered by further extending this technology.
It has previously been demonstrated that increasing the length of the fatty acid side chain gives rise to a longer half-life as observed with insulin detemir,30 insulin degludec23 and oral insulin 338.11 15 For illustrative purposes, the pharmacokinetic profile of insulin icodec has been compared with these acylated basal insulin analogs in dogs (online supplemental figure 2), where an increase in half-life was observed going from C14 to C16, C18 and C20 fatty acids, reflecting the increasing albumin binding affinity. For insulin detemir and insulin degludec, the fatty acid side chain also contributes to the formation of larger soluble self-association forms: dihexamers for insulin detemir and multihexamers for insulin degludec, which occur in the subcutis at the injection site. However, insulin icodec was intentionally formulated not to promote the formation of multimers at the injection site. For a protein drug, multimers with an absorption time longer than a week may not be desirable due to the risk of reduced bioavailability. Instead, for insulin icodec, a reservoir is formed by the albumin-bound depot that is widely distributed throughout the circulation and in interstitial compartments. This albumin-binding mechanism for extending the action of drugs is well established for acylated insulin or GLP-1RAs31 32 and for small molecules and for endogenous hormones such as IGF-1, thyroid and growth hormones that bind to albumin or other protein carriers ensuring a very natural way of prolonging their effect.33 34
Strong binding to albumin, however, was not sufficient to achieve a half-life suitable for once-weekly dosing. Therefore, amino acid modifications were introduced to lower IR affinity and subsequently IR mediated clearance (B16H, A14E and B25H). These modifications are additionally known to increase solubility, thereby helping to achieve a stable U700 formulation.11 The same substitutions also reduce susceptibility to enzymatic degradation11 15 and potentially non-IR mediated clearance, further stabilizing the insulin icodec molecule once administered into the body.
The reduced IR binding affinity of insulin icodec contributes to the long half-life by reducing the clearance rate, since insulin clearance occurs primarily via IR-mediated internalization. Importantly, the low IR affinity does not translate into reduced potency but rather results in a slow rate of insulin action since the low receptor affinity means it will take longer for each insulin molecule to initiate receptor activation before it can be cleared. Thus, insulin icodec remains fully potent, but much slower and longer acting compared with native HI. In addition, this slow initiation of insulin action due to the low IR binding affinity importantly contributes to the safe administration of large insulin icodec doses in one subcutaneous injection by preventing a quick glucose-lowering effect.
Furthermore, as demonstrated in a panel of in vitro systems, despite the low IR binding affinity, it can be seen that insulin icodec is a fully efficacious insulin analog, binding to IRs to activate the same signaling pathways leading to the same full metabolic effects as HI (figure 1, tables 1 and 2). Insulin icodec does not demonstrate increased binding to the IGF-1R, and there is no increased mitogenic potency (figure 1 and table 2). This indicates that insulin icodec has retained the balance between metabolic and mitogenic effects, similar to HI. Furthermore, clinical phase I and II trials indicate that the potency of insulin icodec is similar to other basal insulin products (insulin degludec and insulin glargine U100).26 35
Pharmacokinetic modelling of serum insulin icodec trough concentrations during initiation of once-weekly dosing (figure 2A) demonstrates the build-up of an albumin-bound depot. After 3–4 weekly injections, steady state is reached in the majority of individuals at which time the rate of clearance essentially matches the administered dose. Figure 3 provides a schematic illustration of the sequence of events leading to steady state. With the first injection (panel A1), a week’s dose of insulin icodec is administered into the subcutis, where hexamers dissociate into monomers that are absorbed into the circulation, binding to albumin. A fraction of insulin icodec reaches target tissues, resulting in low glucose-lowering effect, which gradually increases during the week (panel B; first week). The strong albumin binding and low IR affinity (slow initiation of insulin action) ensure that despite the large weekly dose, there is no immediate large glucose-lowering effect. A significant amount of albumin-bound insulin icodec is still present at the end of the first week (panel A2; panel B, label 2). Following the second weekly injection, there is further build-up of albumin-bound insulin icodec and glucose-lowering effect (panel A3; panel B, label 3), which continues with the subsequent weekly injections until the full response is achieved defined as steady state (panel A4; panel B, label 4). At this time, the glucose-lowering effect for insulin icodec over the week is similar to the equivalent once-daily basal insulin glargine U100 dose (figure 3B).

{kind=link}
{kind=link}
{kind=link}
Schematic depiction of build-up to steady state and mechanism of action of insulin icodec. (A) Distribution of insulin icodec (light blue) bound to albumin (grey) in the different compartments over time from initiation of once-weekly dosing. Chart 1: distribution of insulin icodec after the first injection, with the majority of insulin icodec in the subcutis and a small proportion absorbed into the blood. Chart 2: day 7, prior to the second injection, showing that there is still insulin icodec distributed prior to the next injection. Charts 3–4: showing the gradual build-up of insulin icodec exposure towards steady state. (B) Conceptual model showing glucose-lowering effect over time from initiation of once-weekly dosing of insulin icodec and once-daily dosing of insulin glargine U100 (at comparable dose levels). Blue curve: insulin icodec; grey curve: insulin glargine U100. Orange labels refer to charts 1–4 in panel A. AU, arbitrary units.
Results from the clinical pharmacology trial support the model illustrated in figure 3. As shown in figure 2B, the geometric mean half-life was 196 hours, that is, just longer than 1 week. As a consequence of the long half-life, insulin icodec builds up during the first weeks of once-weekly dosing until reaching a plateau. The median tmax was 16 hours, and a peak to trough difference could be observed when measured as total insulin icodec (bound and unbound to albumin). However, the mean glucose-lowering effect of insulin icodec demonstrated a close to even distribution over the entire dosing interval of 1 week (figure 2C).
The maximum serum concentration of insulin icodec at steady state (<500 nmol/L; figure 2B) is substantially lower than the serum albumin concentration (600 000 nmol/L), and each albumin molecule has at least four high affinity fatty acid binding sites and additional intermediate affinity binding sites.36 37 Thus, even in case of higher concentrations of insulin icodec, there is >2000-fold excess of binding sites in the circulating albumin pool compared with the insulin icodec serum concentration. Therefore, displacement of insulin icodec from albumin due to albuminuria, intrinsic factors or competitive protein binding will be minimal and unlikely to have any clinically relevant effect on insulin icodec activity.
Insulin therapy is particularly sensitive to variations, both hour to hour and day to day, as too little insulin will result in hyperglycemia while too much can lead to hypoglycemia. It was therefore critical to ensure a flat and steady glucose-lowering effect of insulin icodec from day to day, despite giving 7 days’ worth of insulin in one dose. The modifications engineered into the insulin icodec molecule ensure that at steady state, variations in dosing time or amount would lead to minimal changes in immediate glucose-lowering effect due to the buffering effect of albumin binding as well as the slow initiation of IR activation. Indeed, in a phase II trial insulin icodec reduced HbA1c and fasting plasma glucose in people with type 2 diabetes to the same extent as once-daily insulin glargine U100 at comparable frequency of level 2 and level 3 hypoglycemia with considerably fewer overall injections.35 Furthermore, in a phase II trial to investigate switching from once-daily basal insulin to once-weekly insulin icodec, the use of an initial 100% loading dose of insulin icodec (first injection only), equivalent to twice the weekly dose was well tolerated, reflecting the advantage of the strong albumin binding and slow rate of insulin icodec action under these conditions.38 This is clearly illustrated in figure 3B, where it can be seen that during the first week, despite the administration of a full dose of insulin icodec, the model-predicted pharmacodynamic effect is considerably less, suggesting that it should be safe to add a loading dose as was investigated in the phase II trial.38
In conclusion, the molecular modifications introduced into insulin icodec make it a basal insulin with properties suitable for once-weekly dosing. The optimized features of strong albumin binding with reduced IR affinity leading to slow IR-mediated clearance contribute towards the formation of an essentially inactive albumin-bound depot, which provides continuous and slow initiation of insulin action. This mechanism is key to achieving safe and efficacious basal glucose lowering35 38 over an entire week with once-weekly dosing of insulin icodec, providing the additional convenience of considerably fewer injections compared with a once-daily basal insulin.
Data availability statement
For the non-clinical studies, data associated with this work are present in the paper. Data and reagents may be made available, on reasonable request, from Novo Nordisk A/S under a materials transfer agreement. For the clinical trial, individual participant data will be shared in data sets in a deidentified/anonymized format. Datasets from Novo Nordisk sponsored clinical research completed after 2001 for product indications approved in both the EU and USA will be shared. Trial protocol and redacted Clinical Trial Report will be available according to Novo Nordisk data sharing commitments. The data will be available permanently after research completion and approval of product and product use in both EU and USA. Data will be shared with bona fide researchers submitting a research proposal requesting access to data. Data will be shared for analyses as approved by the Independent Review Board (IRB) according to the IRB Charter (see novonordisk-trials.com). Data will be available through an access request proposal form, and the access criteria can be found at novonordisk-trials.com. The data will be made available on a specialized SAS data platform.
Ethics statements
Acknowledgments
Technical assistance was provided by Annette F Bjerre, Marianne B Jensen, Pia Jensen, Lenette S Jørgensen, Anette Kirstine Lauridsen, Gitte Norup, Jette E Svendsen and Lene Walander. The authors would like to thank the principal investigator of the clinical pharmacology trial, Ulrike Hövelmann, MD, Profil, Neuss, Germany and all other investigators at Profil for conducting the clinical pharmacology trial, which was sponsored by Novo Nordisk. Medical writing support was provided by Carsten Roepstorff, PhD, CR Pharma Consult, Copenhagen, Denmark funded by Novo Nordisk.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Deceased PM deceased.
Contributors EN contributed with study design, data acquisition, data analysis, critical manuscript revision and final manuscript approval. LP contributed with study conception, design, data acquisition, data analysis, critical manuscript revision and final manuscript approval. TG, BFH, AL, TÅP and CES contributed with data acquisition, data analysis, critical manuscript revision and final manuscript approval. FH and TK contributed with study conception, design, critical manuscript revision and final manuscript approval. NRK contributed with clinical study conception, data analysis, critical manuscript revision and final manuscript approval. KL contributed with clinical study conception, design and data analysis, critical manuscript revision and final manuscript approval. PM contributed with study conception, design and critical manuscript revision. RR-M contributed with clinical data acquisition, data analysis, critical manuscript revision and final manuscript approval. HH contributed with clinical study design, data acquisition, data analysis, critical manuscript revision and final manuscript approval.
Funding This work was funded by Novo Nordisk.
Competing interests All authors are current or past employees of Novo Nordisk A/S, Denmark.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.