Article Text

Abstract
Introduction A potential role for the orphan G protein-coupled receptor, GPR21, in linking immune cell infiltration into tissues and obesity-induced insulin resistance has been proposed, although limited studies in mice are complicated by non-selective deletion of Gpr21.
Research design and methods We hypothesized that a Gpr21-selective knockout mouse model, coupled with type 2 diabetes patient samples, would clarify these issues and enable clear assessment of GPR21 as a potential therapeutic target.
Results High-fat feeding studies in Gpr21−/− mice revealed improved glucose tolerance and modest changes in inflammatory gene expression. Gpr21−/− monocytes and intraperitoneal macrophages had selectively impaired chemotactic responses to monocyte chemoattractant protein (MCP)-1, despite unaltered expression of Ccr2. Further genotypic analysis revealed that chemotactic impairment was due to dysregulated monocyte polarization. Patient samples revealed elevated GPR21 expression in peripheral blood mononuclear cells in type 2 diabetes, which was correlated with both %HbA1c and fasting plasma glucose levels.
Conclusions Collectively, human and mouse data suggest that GPR21 influences both glucose homeostasis and MCP-1/CCL2-CCR2-driven monocyte migration. However, a Gpr21−/− bone marrow transplantation and high-fat feeding study in mice revealed no effect on glucose homeostasis, suggesting that there is no (or limited) overlap in the mechanism involved for monocyte-driven inflammation and glucose homeostasis.
- diabetes mellitus
- type 2
- inflammation
- receptors
- G-protein-coupled
- chemokines
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
GPR21 was proposed as a novel target to treat type 2 diabetes.
Deletion of Gpr21 using traditional methods affected Rabgap1 expression confounding data interpretation and novelty.
What are the new findings?
CRISPR-Cas9 generated Gpr21-deficient mouse displays improved glucose tolerance.
Phenotype not replicated in selectively deleted Gpr21 myeloid cells.
Gpr21−/− monocytes and IP macrophages display selectively impaired chemotaxis.
How might these results change the focus of research or clinical practice?
No, or limited, overlap in the mechanism involved for monocyte-driven inflammation and glucose homeostasis.
Inhibition of GPR21 could yield improvements in both obesity-induced insulin resistance and in diseases in which CCR2-driven inflammation is key, opening up a number of therapeutic indications in which GPR21 antagonists might be effective.
Introduction
Chronic, low-grade inflammation is part of the pathogenesis of obesity-induced insulin resistance that manifests as immune cell infiltration into adipose tissue.1–5 The population of metabolic and immune cells, including adipocytes and macrophages, paralleled by activation of various proinflammatory receptors and signaling pathways, posits a mechanistic link between insulin resistance and inflammation. However, other evidence suggests no causative link, and that improvements in insulin resistance and inflammation occur via separate mechanisms.6 7
Over 30 G protein-coupled receptors (GPCRs) have been implicated in the development and progression of type 2 diabetes (T2D), including orphan receptors.8 However, only the glucagon-like peptide-1 receptor (GLP-1R) has been successfully targeted therapeutically.9 Of these orphan GPCRs, there have been limited studies on GPR21, a receptor widely expressed in metabolically important tissues, including skeletal muscle, brown adipose tissue and hypothalamus.10 11 Notably, GPR21 is expressed at moderate levels in immune cells, including monocytes and various macrophage populations.11
Two independent studies of Gpr21 knockout mice revealed a robust improvement in glucose tolerance and insulin sensitivity after chronic high-fat diet (HFD),11 12 due to reduced tissue inflammation and impaired monocyte and macrophage migration into adipose tissue. Since these promising findings, controversy has arisen around the etiology of the phenotype. A subsequent study suggested that the metabolic improvements were due to disruption of Rabgap1, which encodes for the RAB GTPase activating protein 1 and is the gene within which Gpr21 is nested.13 The authors of this study speculated that insertion of a LacZ-Neo cassette into the Gpr21 gene during the generation of the transgenic mouse disrupted Rabgap1 expression, and it is this effect, rather than disruption of Gpr21, that underlies the metabolic phenotype. This discrepancy in the knockout phenotypes, together with the paucity of other studies on GPR21, demonstrates a need to better understand function of the receptor with improved mouse models and human data.
In an attempt to reconcile differences in the literature, and understand potential roles for GPR21 as a therapeutic target in metabolic diseases, we developed a novel Gpr21 knockout mouse using CRISPR-Cas9 technology for phenotyping in HFD feeding studies, using both whole-body knockout and bone marrow transplant (BMT) models. We also investigated differential expression of GPR21 in control and patients with T2D, and RNA-Seq analysis of, and migration assays with, Gpr21−/− CD11b+ bone marrow monocytes (BMMs).
In contrast with the original Gpr21−/− mouse strain,11 our CRISPR Gpr21−/− mice had unaltered Rabgap1 expression, yet still displayed improved glucose tolerance when fed HFD. Absence of Gpr21 caused a selective reduction in inflammatory markers in adipose tissue and liver, and Gpr21−/− CD11b+ BMMs and intraperitoneal (IP) macrophages exhibited selective impaired chemotactic responses to monocyte chemoattractant protein-1 (MCP-1; CCL2), despite unaltered CCR2 expression. Further analysis revealed that this impaired inflammatory response is due to dysregulated monocyte polarization.
However, the improved metabolic phenotype of Gpr21−/− was not preserved in the BMT model, suggesting that although GPR21 influences both glucose homeostasis and monocyte function, the two phenotypes are not intrinsically linked. We speculate that due to the compelling effects on monocyte function, GPR21 might be a promising therapeutic target for CCR2-driven inflammatory conditions.
Research design and methods
Generation of Gpr21−/− animals and ethics
CRISPR Gpr21−/− mice (containing a 3.8 kb deletion of exon 2 of Gpr21) were generated following backcrossing onto the C57BL/6J background for more than seven generations (Australian Phenomics Network, Melbourne, Australia). Guide RNA target sites flanking exon ENSMUSE00000450988 of the Gpr21 gene were designed using http://crispr.mit.edu/ (online supplemental table 1). Complementary oligonucleotides of target sites were annealed and cloned into BbsI (NEB) digested plasmid pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene plasmid #42230). Single guide RNAs (sgRNAs) were generated using the HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB) according to the manufacturer’s instructions (online supplemental table 1). RNAs were purified (RNeasy Mini Kit, Qiagen, Germany) and Cas9 mRNA (Sigma-Aldrich, Castle Hill, Australia, 30 ng/µL) and sgRNAs (15 ng/µL) were microinjected into the cytoplasm of C57BL/6J zygotes at the pronuclei stage. Zygotes were transferred into the uterus of pseudo pregnant F1 females. Deltagen mice were obtained from Deltagen (USA) as per previous works.11 12
Supplemental material
Animal studies were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and study protocols reviewed and approved by the Monash University (Clayton, VIC, Australia), the Monash University Research Ethics Committee (Clayton, VIC, Australia), Baker Heart and Diabetes Institute (Melbourne, VIC, Australia) and the Alfred Medical Research and Education Precinct Ethics Committee (Melbourne, VIC, Australia). Breeding and experimental procedures of these animals were in accordance with MARP/2015/012/BC, MARP/2015/0140/Exp/Cowley/MA, MIPS/2016/15/(DARREN RIDDY) and 5477HK. To avoid hormonal effects, only male mice were used for experiments. Mice were housed in a 12-hour light, 12-hour dark cycle with food and water ad libitum. For feeding studies, mice were fed either normal chow (NC) (8.9% energy from fat; Specialty Feeds, Glen Forrest, WA, Australia) or HFD (60% energy from fat; SF16-048, Specialty Feeds,). Diets started at week 1 of the study. For genotyping, the primers (Gene Works, Melbourne, Australia) used are described in online supplemental table 2.
Human ethics
Collection and use of human blood samples was conducted according to the guidelines and approval of the Monash University (Clayton, Australia) and the Monash University Research Ethics Committee (Parkville, Australia; 2021-21414-57552). Blood was obtained from donation by vein puncture of human volunteers from the Victorian Blood Donor Registry, or from buffy coat preparations from the Australian Red Cross blood service (Melbourne, Australia, agreement number 18-08VlC-11). Blood was collected by written consent for the purposes of research. Adipose tissue and peripheral blood mononuclear cell (PBMC) clinical samples used were obtained from two studies described in previous works (142016; NCT02368704) and (152019; NCT02671864). Both studies were conducted in accordance with the principles of the Declaration of Helsinki, with informed consent.
Bone marrow isolation and transplantation
Bone marrow (BM) was isolated from the femur and tibia of wild-type (n=3) and Gpr21−/− (n=3) mice by flushing of the BM cavity with phosphate buffered saline (PBS). Cells were washed in RPMI and counted using a hematology analyzer Xs-1000i (Sysmex, Germany). Cells (~3×106 cells) were injected into the tail vein of 8-week-old male irradiated (2×5.5 Gy) wild-type C57BL/6 J mice. Mice were allowed 5 weeks for reconstitution of donor BM and recovery.
Mouse and human cell isolations and function assays
BMMs were isolated as described in previous work.6 CD11b+ monocytes were isolated by negative selection and magnetic bead separation method (Miltenyi Biotech, Germany) according to manufacturer’s instructions. IP macrophages were obtained by flushing the IP space with 0.9% PBS. Human CD14+ PBMCs were isolated as described in previous work.16 Ex vivo migration of human CD14+, mouse CD11b+ BMMs and IP macrophages were performed as described previously.16 Monocyte adhesion and shape change polarization assays were performed as described in previous works17 18 and use of F-actin staining.
Ex vivo migration of CD11b+ BM monocytes and IP macrophages measured by chemotaxis
Chemotaxis assays were performed using HTS-transwell inserts (Sigma-Aldrich, Castle Hill). A volume of 150 µL of chemoattractant (monocyte chemoattractant protein-1; MCP-1, monocyte chemoattractant protein-2; MCP-2, monocyte chemoattractant protein-3; MCP-3, monocyte inhibitory protein-1α; MIP1α) in serum-free growth medium was added to the bottom chamber of the insert. In the top chamber, 50,000 cells resuspended in 50 µL serum-free growth medium were added. A negative control using vehicle and positive control using 10% fetal bovine serum (FBS; replicate wells) were included in each assay. Once the samples were prepared the plates were incubated for 3 hours, at 37°C with 5% CO2. Following the incubation, transwells were removed and the plates dried before fixing the cells with formalin solution containing Hoechst 33258 (Sigma-Aldrich) for nuclei staining. Wells were imaged using an InCell Analyzer 2000 (GE Healthcare, Little Chalfont) and number of cells quantified using Image J V.1.50b (NIH, US).
Metabolic parameters
MRI, oral glucose (OGTT), and IP insulin tolerance tests (IPITT) were performed as described previously.6 Plasma insulin concentrations were measured using a Mouse Ultrasensitive Insulin ELISA kit (ALPCO, Salem, New Hampshire, USA) according to manufacturer’s instructions.
Tissue processing and FACS analysis
Epididymal fat pads were rinsed in PBS and minced in FACS buffer (PBS containing 1% bovine serum albumin (BSA)). Adipocytes and stromal vascular cells were prepared from collagenase-digested adipose tissue. In brief, tissue homogenates were incubated in 1 mL PBS supplemented with 0.5% BSA and 3 mL collagenase II digest solution using a rotational shaker for 20 min at 37°C. All samples were washed twice with PBS and passed through a 70 µm filter. The suspension was centrifuged at 500×g for 10 min at 4°C and resuspended in FACS buffer. FACS analysis of stromal vascular fraction (SVF) for immune cell content were performed using fluorescently conjugated antibodies to various markers, including PB-CD45, APC-CD115, Percp-Gr1, APCCy7-CD3, FITC-CD19, PECy7-F4/80, G05-CD206, PE-NK1.1, and Percp-CD11c (Australian Biosearch, Wangara, WA, Australia). For mouse CD11b+ BM monocytes, cells were isolated from both wild-type and Gpr21−/− animals as described above. Cell populations were characterized using fluorescently conjugated antibodies to Ly6C, Ccr2 and Cx3cr1 (Australian Biosearch, Wangara, WA, Australia). Content was measured by use of a BD FACS LSRFortessa (BD Biosciences, Singapore). Data were analyzed using FlowJo V.10 (LLC, Ashland, Oregon, USA). All data are expressed as % cell population gated from CD45 + cells. Whole blood and liver were prepared as above but in the absence of collagenase.
Gene expression and RNA sequencing
Knockdown of GPR21 in CD14+ PBMCs was achieved using lentiviral shRNA using scrambled or GPR21 specific sequence with a multiplicity of infection (MOI) of 10 (with ~60% efficiency). Cells were sorted by selecting sytox red-/GFP+ cells. mRNA extraction and gene expression analysis were performed as described in previous work.16 Primers (Gene Works, Melbourne, Australia) used for the study are described in online supplemental table 3.
Mouse CD11b+ RNA was isolated from Trizol homogenates using the Direct-zol RNA MiniPrep Kit (Zymo Research, California, USA). RNA depletion and library construction were performed using NEBNext rRNA Depletion Kit and NEBNext UltraTM Directional RNA Library Prep Kit for Illumina (NEB, USA). Library QC was performed by MultiNA Bioanalyzer (Shimadzu Japan) and pooled to equimolar concentration. Libraries underwent Illumina single read sequencing (AGRF, Melbourne, Australia) using HiSeq v4 reagents to generate 100 bp reads.
Data analysis and bioinformatics
Chemotaxis, FACS (fluorescently conjugated antibodies shown in online supplemental table 4) and gene expression analysis were performed as described in previous work.16 Polarization data are expressed as the percentage of monocytes with >1.5-pixel distance. Bioinformatic analysis were performed as described in previous works.19–25 Ternary expression diagrams were generated using the ggtern package and heatmaps were generated using the gplots package in R. Experiments were analyzed using FlowJo V.10 (LLC, Ashland, Oregon, USA) and Prism V.9.0a (GraphPad Software, San Diego, California, USA).
Statistical analysis
In all cases, data are shown and mean+SEM, with the number of replicates indicated within the figure legend. In most cases, statistical significance was deemed by two-way analysis of variance (ANOVA) with Tukey’s post hoc multiple comparison test compared with wild-type animals fed NC. Rabgap1 expression was deemed significant by multiple t-test with Holm-Sidak multiple comparison test, compared with wild-type. Gene expression data were deemed significant using one-way ANOVA with Dunnett’s multiple comparison test compared with wild-type NC (*) or HFD (#).
Results
Previous Gpr21−/− mouse studies have been potentially confounded by effects on Rabgap1, the gene in which Gpr21 is nested.11–13 Using CRISPR-Cas9 technology, we developed a new knockout, termed CRISPR Gpr21−/−. Figure 1A compares the strategies used to generate this mouse compared with the original Gpr21−/− knockout mouse.11 12 We confirm that Rabgap1 is significantly altered in metabolic tissues and immune cells of the original knockout mouse, specifically downstream of Gpr21 (figure 1B–D), while deletion of Gpr21 in our new model did not significantly alter Rabgap1 expression (figure 1E–G).
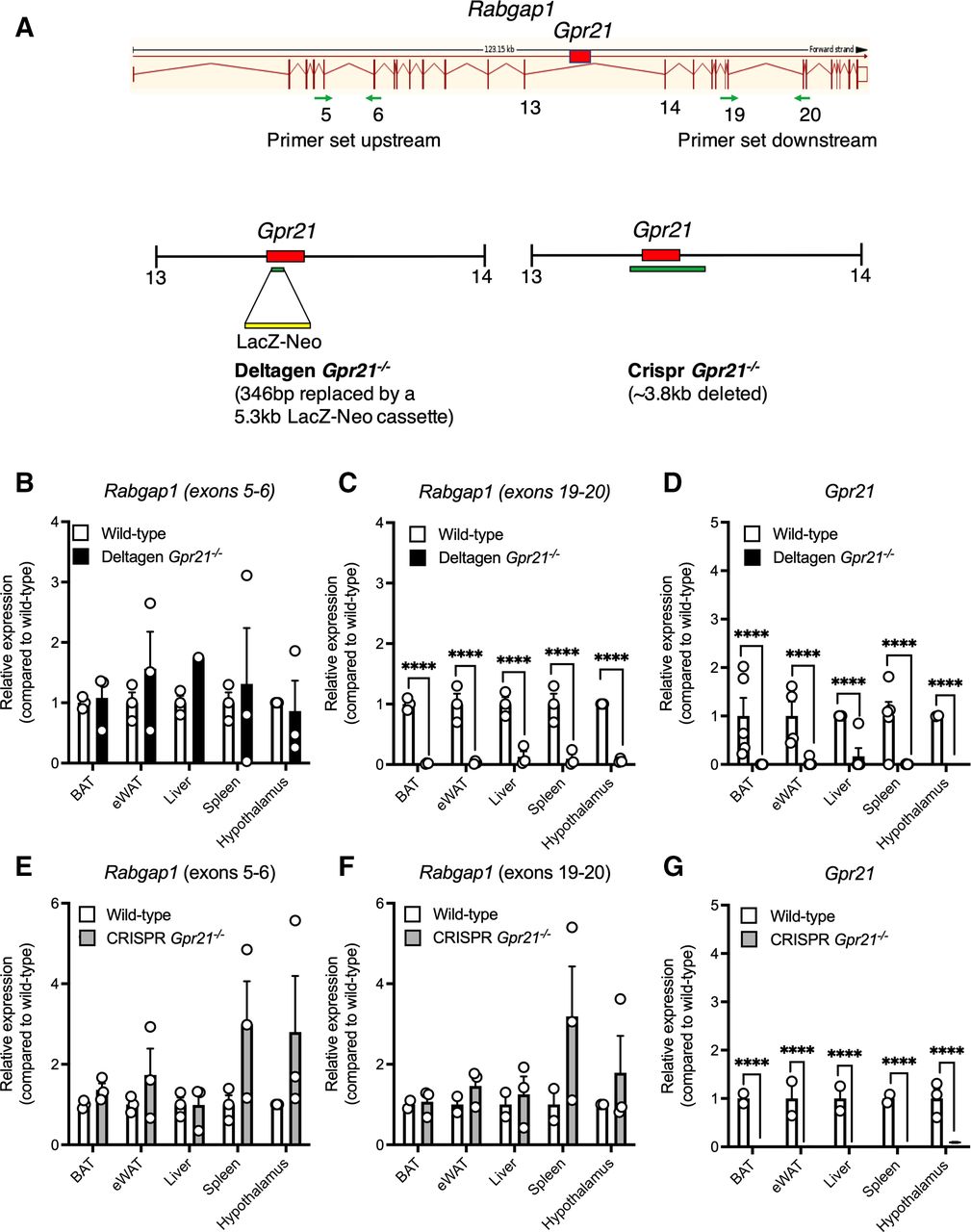
Gpr21 deletion does not affect Rabgap1 expression. (A) Comparison of the regions deleted in the published Deltagen and the CRISPR knockout model used within this study. Relative expression of (B and E) Rabgap1 measured upstream of Gpr21 (exons 5–6), (C and F) Rabgap1 measured downstream of Gpr21 (exons 19–20), and (D and G) Gpr21 in various tissues from wild-type (white bars), Deltagen (black bars) and CRISPR (gray bars) Gpr21−/− animals, respectively. All data are presented as mean+SEM (n=3–4). Multiple t-tests with Holm-Sidak multiple comparison test, ****p<0.0001, compared with wild type. WAT, white adipose tissue.
Having established a clean genetic knockout of Gpr21, we explored the suitability of this new model to study the effects of mice fed HFD. First, we noted no significant difference in body weight of Gpr21−/− mice compared with wild-type littermate controls prior to placing mice on their respective diets (online supplemental figure 1a, n=6–12). After the 16-week feeding period, wild-type controls fed HFD displayed the expected increased body weight compared with NC fed animals (figure 2A,B, n=6–12). Interestingly, Gpr21−/− animals gained less weight on a HFD compared with control mice and, over the duration of the experiment, gained the same amount of weight irrespective of whether they consumed HFD or NC (figure 2B, n=6–12). Although no genotype difference in glucose tolerance was evident after 6 weeks of HFD (online supplemental figure 1a–d, n=6–12), OGTTs after 12 weeks revealed an improvement in glucose handling in Gpr21−/− animals fed HFD compared with controls (figure 2C,D, n=6–12). However, fasting plasma glucose levels appeared not to be influenced by Gpr21 deletion (figure 2E, n=6–12).

Whole-body deletion of Gpr21 reveals improved glucose handling. Changes in (A) body weight (BW) (g), (B) BW % of wild-type and Gpr21−/− mice on normal chow (NC) and high-fat diet (HFD). Week 12 (C) oral glucose tolerance test (OGTT; 3 g/kg lean), (D) area under the curve (AUC), (E) fasting glycemia, relative gene expression of inflammatory and energy expenditure markers in (F) epididymal white adipose tissue (WAT) and (G) liver, as measured by qPCR (n/d=not detected). All data are presented as mean+SEM (n=6–12, unless otherwise stated). Statistical significance was determined by two-way analysis of variance with Tukey’s multiple comparison test compared with wild-type NC, with *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001 deemed significant and compared with wild-type HFD with #p<0.05, ##p<0.01, ###p<0.001, and ####p<0.0001 deemed significant.
At the conclusion of the study, key metabolic tissues were collected and analyzed for changes in the expression of key inflammatory genes. HFD promoted the expression of Ccr2, Ccl2, Il1β, and Nlrp3 in the epididymal WAT (eWAT) and liver of wild-type animals, which was in part lower in the Gpr21−/− mice (figure 2F,G, n=6–12). However, histologic analysis revealed no genotypic differences in lipid droplet number and size (liver) and adipocyte area and circularity (eWAT; online supplemental figure 1e–f).
Given the reduction in Ccr2 expression in eWAT and liver, we explored the potential role of GPR21 in immune cells, specifically monocytes. However, before investigating any potential functional differences, we profiled BMMs isolated from wild-type and Gpr21−/− animals by FACS to check that the isolation procedure yielded similar populations of cells. Markers for phagocytosis and proinflammatory monocytes (CD11b+ Ly6Chigh CCR2high CX3CR1low), proinflammatory (CD11b+ Ly6Cmedium CCR2high CX3CR1low), and patrolling tissue repair anti-inflammatory monocytes (CD11b+ Ly6Clow CCR2low CX3CR1high26) revealed no genotypic differences between the populations isolated (figure 3A,B, n=5). MCP-1 (1–1000 ng/mL) robustly stimulated chemotaxis in wild-type CD11b+ monocytes with a classical bell-shaped curve, a response that was significantly blunted in Gpr21−/− CD11b+ monocytes (figure 3C, n=4–9), despite unchanged cell-surface expression of Ccr2 (figure 3D, n=4). The effect on MCP-1 function was highly specific, as Gpr21−/− CD11b+ monocytes responded to chemotactic gradients of MCP-3 and macrophage inflammatory protein (MIP)-1α in an identical manner to those isolated from wild-type animals, ruling out generalized defects in chemotaxis (figure 3E,F, n=3–4). A similar reduction in chemotaxis to MCP-1 was observed in macrophages isolated from the intraperitoneal space of the same animals (figure 3G, n=4–9).

Deletion of Gpr21 does not alter the CD11b+ bone marrow (BM) monocyte population but selectively inhibits monocyte chemoattractant protein-1 (MCP-1) driven monocyte migration. Representative FACS analysis of CD11b+ BM monocytes isolated from (A) wild-type and Gpr21−/− animals as measured by CD11b+ and Ly6C expression and (B) comparative quantification of cell populations. Chemotaxis of (C) CD11b+ BM monocytes isolated from wild-type and Gpr21−/− animals in response to MCP-1. Relative gene expression of Ccr2 in (D) CD11b+ BM monocytes, as measured by FACS. Chemotaxis of CD11b+ BM monocytes isolated from wild-type and Gpr21−/− animals in response to (E) MCP-3 and (F) macrophage inflammatory protein-1α (MIP1α). Chemotaxis of (G) intraperitoneal (IP) macrophages isolated from wild-type and Gpr21−/− animals in response to MCP-1. All data are presented as mean+SEM (n=7, otherwise stated). Statistical significance was determined by Student’s t-test, or two-way analysis of variance with Tukey’s multiple comparison test compared with wild type, with *p<0.05, and **p<0.01 deemed significant.
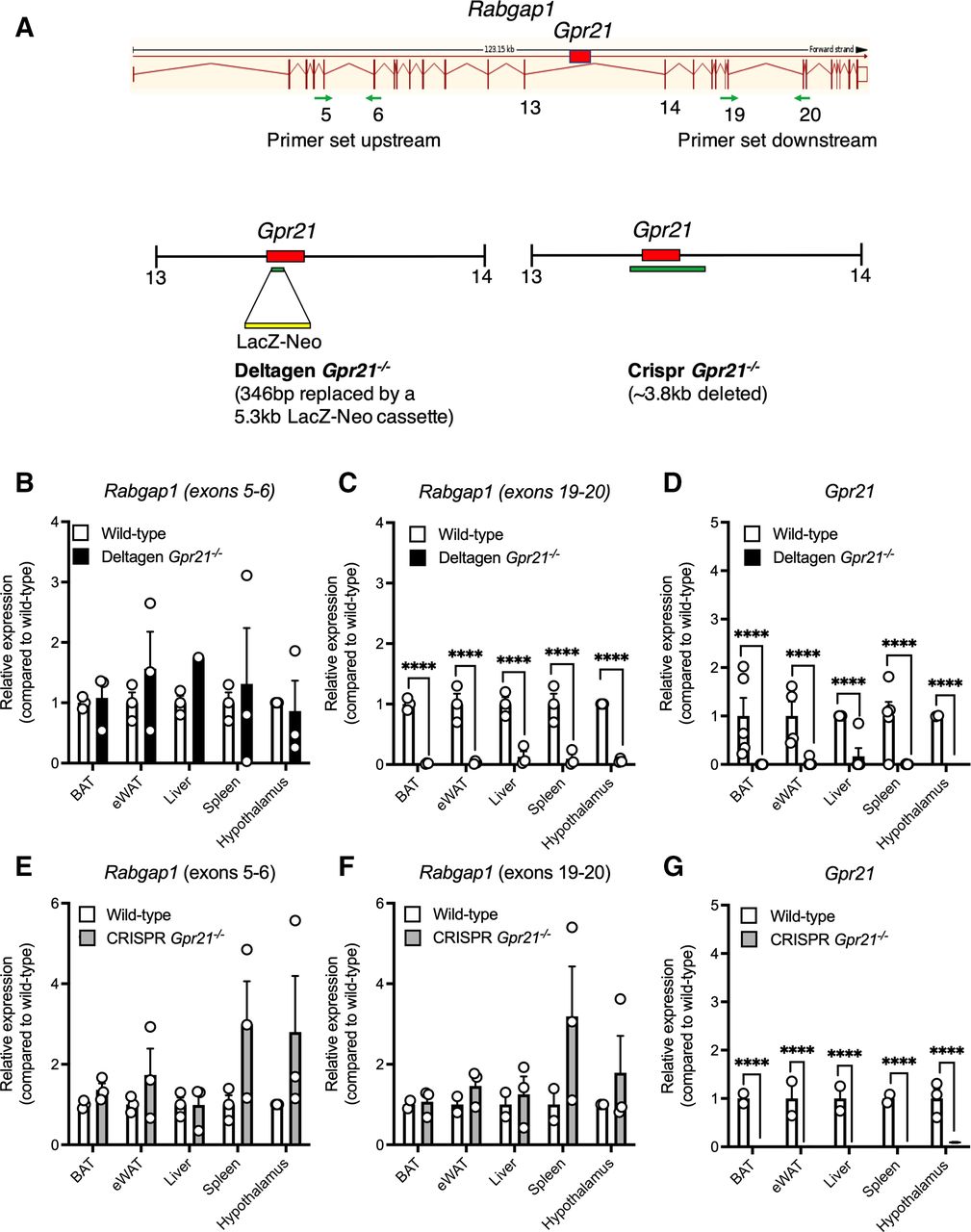
To probe deeper into the role of GPR21 on monocyte function, transcriptomic analysis was performed on CD11b+ BMMs isolated from both wild-type and Gpr21−/− mice. Prior to differential expression analysis, filtering was performed to remove genes with <10 reads. From the initial 48,526 genes, 14,512 were selected for further analysis (online supplemental figure 2a and online supplemental table 5). Analysis revealed 324 differentially expressed genes (DEGs) at a false detection rate (FDR)<0.05, 81<0.01, and 49<0.001 (online supplemental figure 2b). Of the 324 DEGs, 127 were upregulated and 197 downregulated (online supplemental figure 2c); the top 50 regulated genes are shown in a volcano plot in figure 4A and as a heatmap (online supplemental figure 2d and online supplemental tables 6 and 7). Gene set enrichment analysis (GSEA) using sets from Reactome identified an increase in toll-like receptor trafficking, circadian expression, and the NOD, LRR and pyrin domain-containing protein 3 (NLRP3) inflammasome (online supplemental figure 2e), in Gpr21−/− compared with wild-type mice, whereas decreases were observed for collagen formation, extracellular matrix organization and degradation, and nitric oxide signaling (online supplemental figure 2e). GSEA enrichment plots revealed regulation of pathways clearly linked to the phenotype of Gpr21−/− mice, including inflammatory response, monocyte chemotaxis, and GPCR signaling (online supplemental figure 2f,g). Focused analysis of selected genes associated with monocyte adhesion and polarization, including integrins, revealed a downregulation of Itgb3, Itga2b, Itgab2l and CD9 in Gpr21−/− CD11b+ BM compared with wild type (figure 4B). To explore the mechanistic link between these changes and the functional phenotype of Gpr21−/− CD11b+ BMMs, we performed cell adhesion and polarization assays. Wild-type and Gpr21−/− CD11b+ BMMs had a similar capacity to adhere to fibronectin plates following treatment with MCP-1 (100 ng/mL; figure 4C, n=7); however, Gpr21−/− CD11b+ BMMs displayed significantly impaired polarization-induced morphological changes, compared with wild-type cells (figure 4D,E, n=7), which likely underpins the impaired chemotactic response.
Supplemental material

RNA-Seq analysis of CD11b+ bone marrow (BM) monocytes suggests downregulation of integrin expression and impaired monocyte shape change (polarization). Volcano plot (A) of the top 1450 genes (ordered by false detection rate), with genes of interest highlighted (large differences and high significance; top left, downregulated; top right, upregulated). Analysis of integrin expression (B) in RNA-Seq dataset. Time course of (C) adhesion and (D) shape change of CD11b+ BM monocytes isolated from wild-type and Gpr21−/− animals in response to 100 ng/mL monocyte chemoattractant protein-1 (MCP-1), and (E) area under the curve (AUC). All data are presented as mean+SEM (n=5–7, otherwise stated). Statistical significance was determined by multiple t-tests with Holm-Sidak multiple comparison test, ****p<0.0001, compared with wild type, or two-way analysis of variance with Tukey’s multiple comparison test compared with wild type, with *p<0.05 and **p<0.01 deemed significant. Bioinformatics analysis was performed as described in the Research design and methods section.
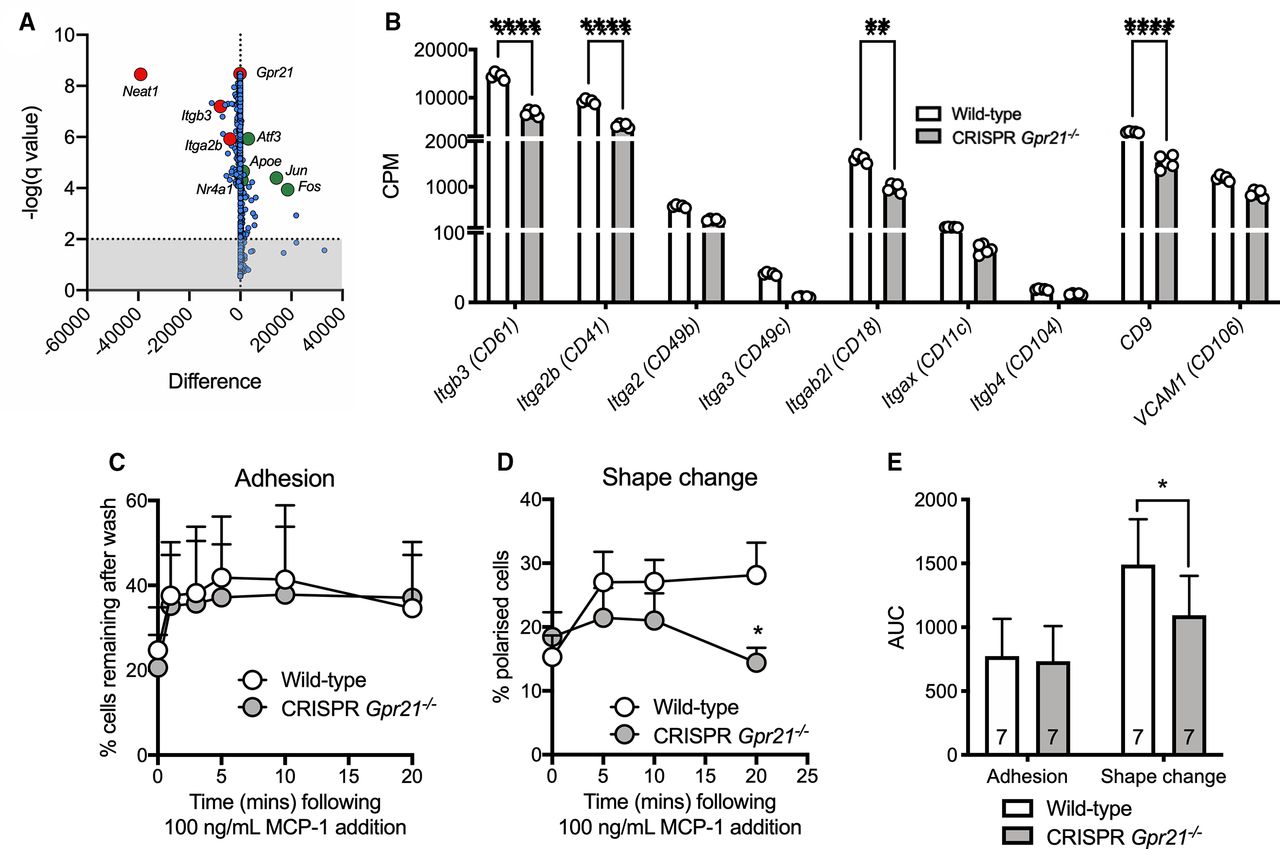
To investigate the translational relevance of GPR21 as a therapeutic target in metabolic disorders, tissue and blood samples from patients with T2D and healthy controls were assessed. mRNA levels for both the proinflammatory MCP-1 (CCL2) and GPR21 were higher in WAT of patients with T2D compared with matched controls (figure 5A,B; n=8–16). Furthermore, there was a significant correlation between WAT GPR21 expression and %HbA1c levels in patients with T2D (figure 5C; *p<0.015, n=24; mean %HbA1c 7.4±1.2;15 2019; NCT02671864). As increased Gpr21 expression in eWAT from mice fed HFD is found in the SVF, but not the adipocyte population (figure 5D; **p<0.007, n=8), we extended patient profiling to CD14+ PBMCs and found that GPR21 expression was significantly higher in CD14+ cells from patients with T2D (figure 5E; n=26–82), which also correlated with fasting plasma glucose (figure 5F; **p<0.005, n=86). Emphasizing a link between MCP-1 and GPR21 in CD14+ PBMCs from healthy blood donors, we observed a correlation between expression of GPR21 and CCR2B, the primary receptor for MCP-1 (figure 5G, *p<0.017, n=8). Finally, to explore a possible functional link between these genes we used a lentiviral-mediated knockdown of GPR21 in CD14+ PBMCs isolated from healthy donors, which resulted in significantly impaired chemotaxis (figure 5H, n=7). Collectively, these clinical data provide evidence linking GPR21 to both glucose homeostasis and regulation of the MCP-1/CCL2-CCR2 axis.

GPR21 is upregulated in white adipose tissue (WAT) and immune cells of patients with type 2 diabetes (T2D). GPR21 (A) and CCL2 (B) mRNA levels are upregulated in WAT of patients with T2D, and GPR21 expression correlates with %HbA1c (C). Expression of mouse Gpr21 is increased in the stromal vascular fraction (SVF) of mice fed high-fat diet (HFD), but not in adipocytes (D). Expression of GPR21 (E) is upregulated in CD14+ peripheral blood mononuclear cells (PBMCs) and correlates with (F) fasting plasma glucose levels in patients with T2D. Expression of (G) GPR21 in PBMCs correlates with CCR2B expression and (H) knockdown by selective GPR21-LV-shRNA reduces monocyte migration in response to 10% FBS. All data are expressed as mean+SEM. Replicates are shown on each panel. Statistical significance was determined by Student’s t-test, one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test compared with wild-type control, or two-way ANOVA with Sidak’s multiple comparison, with *p<0.05 deemed significant. FBS, fetal bovine serum; NC, normal chow.
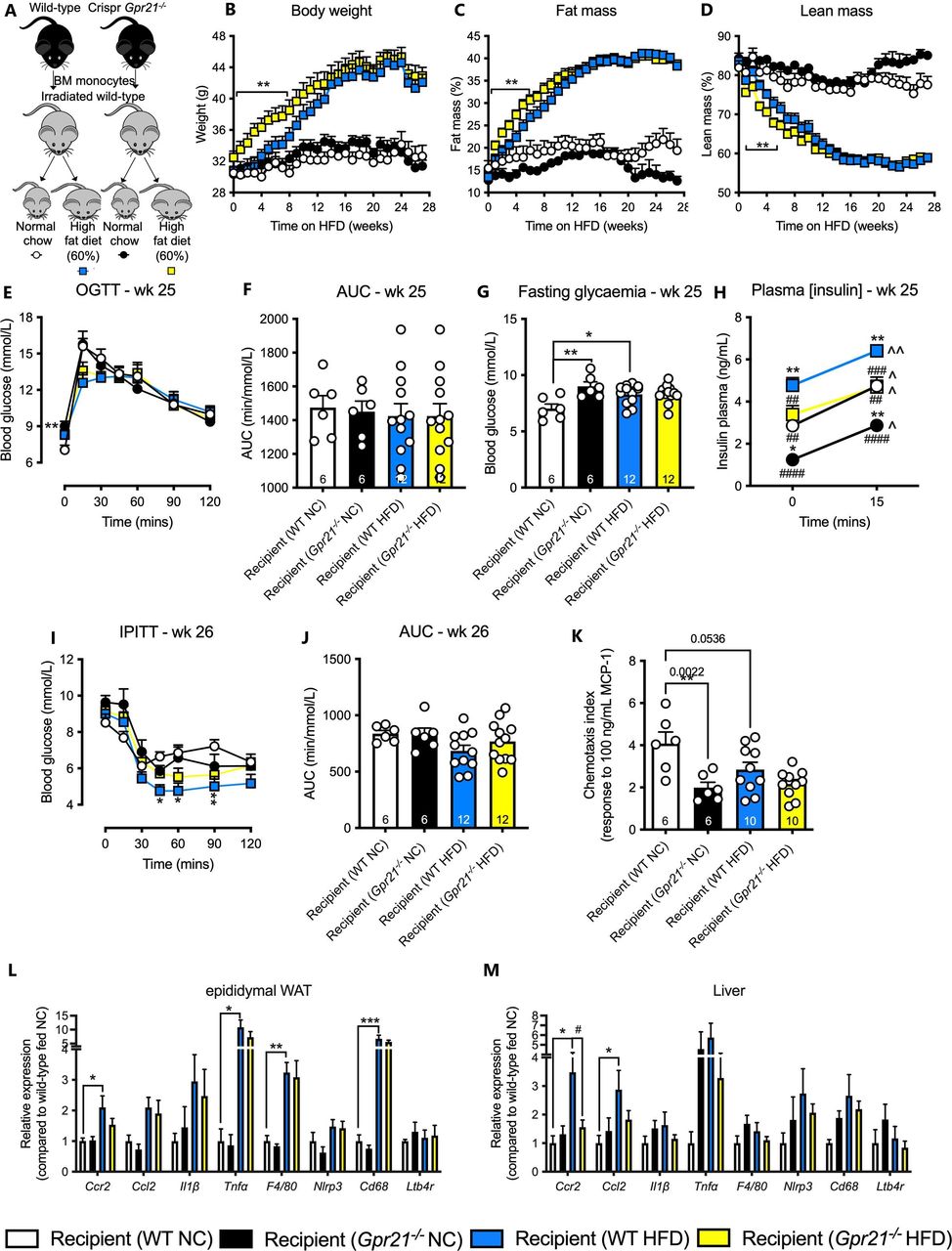
Given that convergent mouse and human data suggest GPR21 both plays a role in glucose homeostasis and is a major regulator of MCP-1/CCR2-driven chemotaxis and inflammation, we sought to determine the extent to which these two phenotypes were linked. To test whether the protective effect on glucose homeostasis was due solely to loss of Gpr21 in cells of the hemopoietic lineage, we performed a bone marrow transplant (BMT; experimental overview figure 6A, n=6–12), followed by HFD feeding and assessments measured as per the whole-body study. All animals placed on HFD increased their body weight and fat mass, and decreased their lean mass (figure 6B–D, n=6–12). Initially, Gpr21−/− recipient animals on HFD gained weight at a faster rate than wild-type controls (figure 6B–D, n=6–12), although this difference was lost after 8 weeks. Although 25 weeks of HFD modestly increased fasting plasma glucose levels in wild-type mice, there was no impairment in glucose handling (figure 6E–G, n=6–12). Gpr21−/− recipient animals on NC also displayed significantly higher fasting plasma glucose than wild-type controls (figure 6G, n=6–12). All groups displayed increased insulin levels in response to glucose challenge, and Gpr21−/− recipient mice had lower levels of plasma insulin both before and during GTTs compared with their respective wild-type controls (both for NC and HFD fed mice; figure 6H, n=11). No improvement in insulin tolerance was observed over a 2-hour time course and as measured by AUC (figure 6I,J, n=6–12). Interestingly, isolated Gpr21−/− CD11b+ BMMs at the end of the study failed to respond to 100 ng/mL MCP-1 compared with wild-type (figure 6K), confirming the chemotactic phenotype described above (figure 2C). Although Gpr21−/− BMT recipient animals did not display improved glucose handling, tissue profiling revealed similar changes in pro-inflammatory gene expression as seen in the whole-body knockout study. HFD increased Ccl2, Ccr2, Tnfα, F4/80, and Cd68 expression in eWAT and liver compared with wild-type animals fed NC; the increased Ccr2 in liver was abrogated in Gpr21−/− recipient animals (figure 6L–M, n=6–12). At a cellular level, HFD significantly elevated the proportion of Ly6Chi monocytes in the SVF of eWAT compared with NC-fed wild-type animals (online supplemental figure 3a–c), but this difference was lost in Gpr21−/− recipient mice. Similarly, on a NC diet, wild-type recipient mice had higher levels of Ly6Clo compared with Ly6Chi monocytes, a difference that was absent in Gpr21−/− recipient animals (online supplemental figure 3d,e). These data confirm that Gpr21−/− recipient mice largely retain the MCP-1/CCR2 regulatory phenotype, but not protective effects on glucose homeostasis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Bone marrow transplant (BMT) study reveals no effect on metabolic phenotype. (A) Schematic diagram of BMT study. Changes in (B) body weight (BW), (C) % fat mass, and (D) % lean mass. Week 25 (E) oral glucose tolerance test (OGTT; 3 g/kg lean), (F) area under the curve (AUC), (G) fasting glycemia, (H) plasma insulin levels (n=6–12). Week 26 (I) intraperitoneal insulin tolerance test (IPITT; 0.6 U/kg body weight for animals on normal chow (NC), or 0.75 U/kg body weight for animals fed high-fat diet (HFD)), (J) AUC. Chemotaxis of (K) CD11b+ bone marrow (BM) monocytes isolated following completion of the BMT study from wild-type (WT) and Gpr21−/− recipient animals in response to 100 ng/mL monocyte chemoattractant protein-1 (MCP-1). Post study relative gene expression of inflammatory and energy expenditure markers in (L) epididymal white adipose tissue (WAT) and (M) liver, as measured by qPCR (n/d=not detected). All data are presented as mean+SEM (n=6–12, unless otherwise stated). Statistical significance was determined by two-way analysis of variance (ANOVA) with Tukey’s multiple comparison test compared with wild-type NC, with *p<0.05, **p<0.01 and ***p<0.001 deemed significant, compared with wild-type HFD, with #p<0.05, ##p<0.01, ###p<0.001 and ####p<0.0001 deemed significant, and compared with t=0 (min), with ˆp<0.05 and ˆˆp<0.01 deemed significant.
Discussion
There is significant debate over GPR21 as a therapeutic target for metabolic diseases. Previous studies of Gpr21−/− mice reported improvements in glucose tolerance and insulin sensitivity in mice fed HFD, putatively driven by reduced levels of metabolic tissue inflammation.11 12 Subsequent studies ascribed this phenotype to modulation of Rabgap1, the gene in which Gpr21 is nested,13 questioning the role of GPR21 in regulating metabolism and/or inflammation.
Current therapies for T2D focus primarily on β-cell dysfunction and insulin resistance. However, the role of chemokine and cytokine-mediated chronic, low-grade inflammation in the pathophysiology of T2D and its complications has gained traction as an alternative area for therapeutic intervention.27 Consumption of high-fat foods and reduced physical activity leads to alterations in immune cell populations. Furthermore, a significant increase in the expression levels of proinflammatory mediators, including interleukin (IL)-1β, IL-6, IL-10 and MCP-1, is routinely observed in patients with T2D,5 shifting the balance between M2 anti-inflammatory and M1 proinflammatory macrophages. Efforts targeting this increased cytokine release have focused on neutralizing antibodies or anti-inflammatories, including anakinra (IL-1), salsalate (IKKbeta–NF-kappaB inhibition) and TNF antagonists.28 29 This lack of efficacy is exemplified by the failure of canakinumab, an IL-1β-targeted monoclonal antibody, to reduce the incidence of new onset T2D, despite ameliorating levels of high-sensitivity C-reactive protein, and IL-6.30 31 It remains to be seen whether a therapeutic agent focused on an ‘upstream’ regulator of chronic, subclinical inflammation might be more effective than targeting individual inflammatory mediators. Establishing the role of GPR21 in regulating glucose homeostasis and inflammation is thus critical to its evaluation as a potential therapeutic target.
Collectively, our findings suggest that deletion of GPR21 inhibits inflammation caused by high-fat feeding, in accordance with previous studies.11 12 As Rabgap1 expression was preserved in our CRISPR-Cas9 knockout mouse, the effects on inflammation and improvement in glucose homeostasis in the whole-body knockout study are likely due to Gpr21 deletion. This contrasts with the findings of Wang et al,13 who used transcription activator-like effector nucleases (TALEN) technology to generate a Gpr21−/− mouse with unperturbed Rabgap1, and saw no change in glucose handling. However, the report lacked a chronic HFD group and it was only after such a regimen that our study revealed the beneficial effects of Gpr21 deletion. Furthermore, it is not clear to what extent the gene-editing methods employed might contribute to the discrepancy, beyond reported differential editing efficiencies.32 33
Despite the correlation observed in both clinical samples and the whole-body knockout mouse between GPR21, metabolic parameters, and inflammatory markers, the results of the BMT study suggested that effects on glucose metabolism and inflammation were not directly linked, as hematopoietic-specific deletion of GPR21 had no effect on glucose homeostasis. The most parsimonious explanation is that GPR21 regulates both monocyte-driven inflammation and glucose homeostasis, although with limited or no overlap in the mechanisms involved or functional sequelae, consistent both with previous work from our group demonstrating a decoupling of inflammation and insulin resistance6 and the mixed literature surrounding the effects of CCR2 antagonists on glucose homeostasis.34 35
With our Gpr21−/− in vivo metabolic studies, deletion of Gpr21 in cells outside of the hemopoietic lineage is required to see effects on HFD-induced changes in metabolic endpoints. Although Gpr21 has widespread expression, a potential locus for this effect is the hypothalamus, since this tissue displays the highest expression levels of the receptor. In the original Osborn et al11 study, a disconnect in body weight effect between BMT and whole-body knockout studies (similar to that observed herein) prompted the authors to silence Gpr21 in the hypothalamus using lentiviral shRNA, leading to a modest reduction in body weight without changes in glucose homeostasis. This suggests additional, non-hemopoietic roles for GPR21 that contribute to the whole-body knockout phenotype and may enable separation between the observed improvement in glucose homeostasis and their reduced body weight. However, the role of myeloid populations in the brain cannot be excluded and would require further study, for example, with a conditional knockout mouse.
The only major difference between our BMT study and that of Osborn et al11 is the method used to produce the knockout mouse. This suggests that Rabgap1 could play an as yet unidentified role in immune cell function and glucose metabolism (expression was impaired in the original Gpr21−/− mouse). For example, Rabgap1 is involved in the coordination of microtubule and Golgi dynamics during the cell cycle and metaphase to anaphase transition in HeLa cells.36 37 Another explanation is that the BMT itself might protect against an insulin resistant phenotype. In a recent study, a syngeneic BMT of HFD-fed C57BL/6 mice following lethal irradiation, yielded reduced HFD-induced obesity, reduced adipose tissue immune cell infiltration, and decreased insulin secretion when compared with HFD control mice.38 Furthermore, in similar studies using ob/ob mice, reduced adiposity was observed following a BMT compared with non-BMT controls,39 perhaps highlighting a limitation of BMT in metabolic research.
The most compelling data obtained from this study was that deletion of GPR21, in either human and mouse monocytes, significantly decreases inflammatory chemotaxis, most notably and specifically to MCP-1, without a change in the expression of its cognate receptor, CCR2. As migration is a highly complex and tightly regulated process,40 41 we investigated whether changes upstream of monocyte extravasation could have been disrupted. Using confocal imaging techniques, we showed that Gpr21−/− CD11b+ BMMs display delayed polarization in response to MCP-1. To what extent this delay is causative of reduced monocyte migration requires further investigation. However, transcriptomic analysis of Gpr21−/− CD11b+ BMMs revealed downregulation of key genes involved in the adhesion cascade, including Itgb3, Itgax and VCAM, indicative of altered extracellular matrix organization and degradation, that may play a role in the altered chemotactic responses. A comprehensive analysis of these integrin markers at the protein level, as well as monocyte interactions with endothelial cells, would be needed to further elucidate the mechanism(s) by which deletion of Gpr21 regulates monocyte migration to MCP-1.
Intriguingly, the RNA-Seq analysis identified a number of other genes of interest. Both Jun and Fos were significantly upregulated in the Gpr21−/− CD11b+ BMMs. Differentiation of human PBMCs by M-CSF results in anti-inflammatory macrophages42 and an upregulation of both Jun and Fos,43 allowing us to speculate that Gpr21−/− CD11b+ BMMs might display an M2, anti-inflammatory-like phenotype and that GPR21 could be involved in suppressing chemokine expression and signaling, which correlates with the reduced effect of MCP-1. Furthermore, these data may indicate GPR21 as a potential target for the treatment of inflammatory diseases where MCP-1 and/or CCR2 has pathogenic roles, including atherosclerosis.
In summary, using a new Gpr21 knockout mouse model, we demonstrate that whole-body deletion of the receptor ameliorates glucose intolerance induced by HFD, which is accompanied by a normalization of selected inflammatory markers. We also show that GPR21 regulates inflammatory chemotaxis in both mouse and human monocytes, likely due to altered monocyte polarization and adhesion/integrin expression and function, although a BMT study suggests that these two phenotypes are likely not linked. Finally, we describe significantly higher expression of GPR21 in patients with T2D, which appears to correlate with CCR2 expression and function. Collectively, these data suggest that an inhibitor of GPR21 could yield improvements in both obesity-induced insulin resistance and in diseases in which CCR2-driven inflammation is a cardinal feature, opening up a number of therapeutic indications in which GPR21 antagonists might be effective.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Acknowledgments
AC, PMS, and MAF are/were Senior Principal Research Fellows of the Australian National Health and Medical Research Council. AJM is a CSL Centenary Fellow. Thanks to Mr Cameron Nowell for developing the scripts for the adhesion and polarization analysis (Drug Discovery Biology, Monash Institute of Pharmaceutical Sciences, Monash University, Australia).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DMR, HLK and AJM contributed equally.
Contributors DMR: Conceptualization, formal analysis, investigation, writing—original draft, supervision, project administration. HLK: Conceptualization, formal analysis, investigation, writing—original draft. AJM: Conceptualization, formal analysis, investigation, writing—original draft. SB-G: Investigation, formal analysis. RDlFG: Investigation, formal analysis. JM: Investigation, formal analysis. MZ: Formal analysis, data curation. SF: Resources. TLP: Investigation. ND: Investigation. PR: Investigation. AE-O: Data Curation. J-FG: Resources. NV: Resources. WNC: Writing—review and editing, funding acquisition. AC: Writing—review and editing, funding acquisition. PMS: Writing—review and editing, funding acquisition. RJS: Writing—review and editing, funding acquisition. MAF: Conceptualization, writing—review and editing. PD: Conceptualization, writing—review and editing, funding acquisition. CJL: Conceptualization, formal analysis, writing—review and editing, supervision, project administration, funding acquisition. DMR and CJL are responsible for the overall content as the guarantor.
Funding The study was funded by Servier.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.