
Abstract
Aims/hypothesis
In previous studies we have shown that extravasated, modified LDL is associated with pericyte loss, an early feature of diabetic retinopathy (DR). Here we sought to determine detailed mechanisms of this LDL-induced pericyte loss.
Methods
Human retinal capillary pericytes (HRCP) were exposed to ‘highly-oxidised glycated’ LDL (HOG-LDL) (a model of extravasated and modified LDL) and to 4-hydroxynonenal or 7-ketocholesterol (components of oxidised LDL), or to native LDL for 1 to 24 h with or without 1 h of pretreatment with inhibitors of the following: (1) the scavenger receptor (polyinosinic acid); (2) oxidative stress (N-acetyl cysteine); (3) endoplasmic reticulum (ER) stress (4-phenyl butyric acid); and (4) mitochondrial dysfunction (cyclosporin A). Oxidative stress, ER stress, mitochondrial dysfunction, apoptosis and autophagy were assessed using techniques including western blotting, immunofluorescence, RT-PCR, flow cytometry and TUNEL assay. To assess the relevance of the results in vivo, immunohistochemistry was used to detect the ER stress chaperon, 78 kDa glucose-regulated protein, and the ER sensor, activating transcription factor 6, in retinas from a mouse model of DR that mimics exposure of the retina to elevated glucose and elevated LDL levels, and in retinas from human participants with and without diabetes and DR.
Results
Compared with native LDL, HOG-LDL activated oxidative and ER stress in HRCP, resulting in mitochondrial dysfunction, apoptosis and autophagy. In a mouse model of diabetes and hyperlipidaemia (vs mouse models of either condition alone), retinal ER stress was enhanced. ER stress was also enhanced in diabetic human retina and correlated with the severity of DR.
Conclusions/interpretation
Cell culture, animal, and human data suggest that oxidative stress and ER stress are induced by modified LDL, and are implicated in pericyte loss in DR.
Similar content being viewed by others
Introduction
Pericytes are essential for retinal capillary structure and function, and pericyte loss is an early feature of diabetic retinopathy (DR) [1–4]. Pericyte loss involves apoptosis [3], while the inhibition of apoptosis with caspase inhibitors improves cell viability in models of DR in vivo and in vitro [5, 6]. The specific mechanisms of pericyte loss in DR are unknown. Diabetes enhances glycation and oxidation of proteins, including LDL [7, 8]. In cell culture studies addressing atherogenesis in diabetes, glycated LDL increased cholesteryl ester synthesis and accumulation in human macrophages, accelerating foam cell formation [9]. In vivo, once glycated LDL becomes extravasated in the arterial sub-intimal space, modification is likely to be accelerated. Glycated LDL is more likely than native LDL (N-LDL) to be sequestered, to aggregate and to undergo oxidation, resulting in a heterogeneous product, oxidised and glycated LDL [10], which is recognised by the scavenger receptor [11, 12].
We hypothesised that an analogous process occurs in the retina, i.e. that extravasated, modified plasma lipoproteins contribute to the propagation of DR once retinal capillary leakage is established [13–18]. Using immunohistochemistry, we were the first to show that extravasation and oxidation of LDL occur in the retina in diabetes to an extent proportional to the severity of DR [19]. Extravasated LDL was detectable before the onset of clinical DR, initially in the vicinity of the inner retinal capillaries, i.e. adjacent to pericytes. No such extravasation was detected in healthy, non-diabetic human retinas. Furthermore, we showed that intra-retinal oxidised LDL (Ox-LDL) was also a feature of human DR, again being present in amounts proportional to disease severity, and co-localising with apoptotic cells [19]. In retinas from participants with proliferative DR, Ox-LDL was present throughout all retinal layers, co-localising with macrophages. These data suggest that extravasated, modified LDL may play a key role in the pathogenesis of DR from the earliest stages, and may contribute to pericyte loss.
The present study aimed to investigate mechanisms whereby modified LDL affects human retinal capillary pericytes (HRCP). Possibilities include enhanced oxidative stress, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, apoptosis and autophagy. Oxidative stress has long been considered an initiating factor in diabetic vascular disease [20], and may act via multiple downstream pathways. ER stress is implicated in retinal endothelial cell death and DR in animal models, and its blockade can inhibit progression of DR [21]. Mitochondrial dysfunction is also implicated in pericyte loss induced by hyperglycaemia [22, 23]. While autophagy has been linked to the pathogenesis of diabetes [24, 25], its role in pericyte loss is unexplored.
We used cultured HRCP to determine the effects of in vitro-modified human LDL on these pathways, comparing the effects of N-LDL and ‘highly-oxidised glycated’ LDL (HOG-LDL). To determine mechanisms and to explore the sequence of events, we used inhibitors including: (1) polyinosinic acid (Poly-I), a scavenger receptor inhibitor; (2) N-acetyl cysteine (NAC), an oxidative stress inhibitor; (3) 4-phenyl butyric acid (4-PBA), an ER stress inhibitor; and (4) cyclosporin A (CsA), a mitochondrial dysfunction inhibitor when employed at low micromolar concentrations. Two commercially-available lipid oxidation products, 4-hydroxynonenal (4-HNE) and 7-ketocholesterol (7-KC), were used to confirm findings and to define their utility, when used place of modified LDL, for future studies. To test the relevance of our findings in vivo, and specifically those regarding ER stress, we used immunohistochemistry to detect Ox-LDL and ER stress markers in retinas from a mouse model of combined diabetes and hyperlipidaemia (streptozotocin-induced diabetes with genetically elevated LDL), and in retinas obtained post mortem from human volunteers with or without diabetes and DR.
Methods
The study was approved by the Institutional Review Board at the University of Oklahoma Health Sciences Center (OUHSC) and was conducted according to the principles of the Declaration of Helsinki. Written informed consent was obtained from participants. Animal experiments were approved by the Institutional Animal Care and Use Committee of OUHSC.
LDL preparation, modification and characterisation
Protocols were as previously described [26]. Briefly, human LDL was isolated by sequential ultracentrifugation (350,000 g, densities [d] = 1.019–1.063) of pooled plasma from four to six fasting, healthy volunteers. N-LDL and glycated LDL were prepared by incubating LDL without and with freshly prepared 50 mmol/l glucose (72 h, 37°C) under anti-oxidant conditions, i.e. 1 mmol/l N,N-bis[2- (bis[carboxymethyl]-amino)ethyl]glycine (DTPA) with 270 μmol/l EDTA, under nitrogen. HOG-LDL was prepared by oxidising glycated LDL in the presence of 10 μmol/l CuCl2 (24 h, 37°C), followed by repeated dialysis (4°C, 24 h). Protein content in LDL preparations was determined by BCA protein assay (Pierce, Rockford, IL, USA). LDL preparations were further characterised by measuring fluorescence at 360 nm (excitation) and 430 nm (emission) (Fluorimeter IV; Gilford, Oberlin, OH, USA), performing agarose gel electrophoresis (Paragon Lipo Gel; Beckman, Fullerton, CA, USA) and measuring absorbance (234 nm; DU 650 Spectrophotometer; Beckman). Preparations were stored in the dark under nitrogen at 4°C in the presence of 270 μmol/l EDTA and were used within 6 weeks. Experiments were repeated using different LDL preparations.
HRCP cell culture
HRCP (Cambrex, Walkersville, MD, USA) were cultured (37°C, under 5% CO2 [vol./vol.]) in endothelial basal medium 2 (Lonza, Allendale, NJ, USA) supplemented with endothelial growth medium 2 from a kit (Single Quots; Lonza). Cells (passages 3–9) at 85% confluence were treated in serum-free medium (SFM) for 18 to 24 h to induce quiescence, followed by treatment with N-LDL, HOG-LDL, 4-HNE or 7-KC at the concentrations and times indicated. In some experiments, pharmacological reagents (Poly-I [50 μg/ml], NAC [100 μmol/l], 4-PBA [0.5 mmol/l] or CsA [2 μmol/l]) were added to media 1 h before adding N-LDL (200 mg protein/l) or HOG-LDL (200 mg protein/l). Each experiment was repeated at least three times.
Cell viability assay
HRCP were seeded into 96-well plates (1 × 104 cells/well). After exposure to experimental conditions for 24 h, cell viability was measured by a cell counting kit assay (CCK-8; Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer’s protocol.
Western blot studies for oxidative stress, ER stress and autophagy
Cells were homogenised with a complete lysis buffer (Roche, Indianapolis, IN, USA). Protein concentrations were determined by BCA protein assay (Pierce). Protein (30 μg) was resolved by SDS-PAGE, then blotted with antibodies against: (1) superoxide dismutase 2 (SOD-2), glutathione peroxidase 1 (GPX-1), protein-bound 3-nitrotyrosine (3-NT), 78 kDa glucose-regulated protein (GRP78), phospho-eukaryotic initiation factor 2α (p-eIF2α) and C/EBP-homologous protein (CHOP) (all from Abcam, Cambridge, MA, USA); and (2) cytochrome C (CYT-C), caspase-3, cleaved poly ADP ribose polymerase (PARP), B-cell lymphoma 2-associated X protein (BAX), B-cell lymphoma 2 (BCL-2), microtubule-associated protein 1 light chain 3 (LC3), autophagy-related homologue 5 (ATG5) and beclin-1 (all from Cell Signaling Technology, Danvers, MA, USA). Blots were subsequently stripped and re-blotted with antibody against β-actin (Abcam) to assess protein load.
Measurement of reactive oxygen species
Intracellular reactive oxygen species (ROS) were measured with chlorofluorescein diacetate (Life Technologies, Carlsbad, CA, USA) as per manufacturer’s instructions at numerous time points up to 6 h after treatment with N-LDL or HOG-LDL (200 mg/l).
Immunofluorescence for activating transcription factor 6 (ER stress) and TUNEL assay (apoptosis)
HRCP were seeded and grown to 85% confluence on glass coverslips. After quiescence for 24 and 1 h of pretreatment with Poly-I, NAC, 4-PBA or CsA, cells were treated with HOG-LDL or N-LDL for 24 h. After fixation (4% paraformaldehyde [wt./vol.] for 20 min) and permeabilisation, cells were incubated overnight at 4°C with primary antibody against activating transcription factor 6 (ATF6) (Abcam), followed by a secondary antibody for 1 h. Apoptosis was assessed using an in situ cell death detection kit (Roche) according to the manufacturer’s instructions. Immunofluorescence was visualised under a fluorescence microscope (Nikon, Tokyo, Japan).
Quantitative real-time PCR (ER stress)
RNA was extracted from HRCP (RNeasy Mini Kit; Qiagen, Valencia, CA, USA). Real-time RT-PCR was performed using a cDNA synthesis kit (iScript; Bio-Rad Laboratories, Hercules, CA, USA) and SYBR Green PCR Master Mix (Bio-Rad). mRNA levels of the target genes CHOP (also known as DDIT3) and sXBP-1 (spliced isoform of X-box binding protein 1) were normalised by 18s ribosomal RNA levels. Primer sequences are shown in the electronic supplementary material (ESM) Table 1.
Mitochondrial dysfunction
Mitochondrial membrane potential (Δψm) was determined by flow cytometry using 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)) (DiOC6(3) Detection Kit; Life Technologies), using DiOC6(3) concentrations specific for mitochondria as described [27].
Genetically modified mouse model of hyperlipidaemia
To assess relevance in vivo, we used an animal model combining diabetes and hypercholesterolaemia. We used genetically modified C57B16 mice with double knockout of the gene encoding the LDL receptor (Ldlr −/−) and the apolipoprotein (Apo) B mRNA-editing catalytic polypeptide (enables conversion of ApoB100 to ApoB48) (Apobec1 −/−), along with wild-type controls (Genentech, South San Francisco, CA, USA), as described [28]. Half of the animals of each group were rendered diabetic with streptozotocin as described [28]. At 40 weeks after streptozotocin treatment animals were killed, and eyes were removed, fixed in formalin and then sectioned and immunostained to detect retinal ER stress.
Human retinas
Retinas were obtained postmortem through the National Disease Research Interchange (NDRI) from type 2 diabetic and non-diabetic human donors.
Immunohistochemistry of retinal sections from mice and humans (ER stress)
Retinal immunohistochemistry was performed as described [19]. Briefly, prefix-fixed retinal sections were incubated (overnight, 4°C) with rabbit polyclonal anti-GRP78 antibody, or rabbit polyclonal anti-ATF6 or anti-human ApoB antibodies (Abcam). Anti-rat or anti-rabbit fluorescence-conjugated antibodies (Life Technologies, Carlsbad, CA, USA) were used as secondary antibodies. Immunofluorescence was observed under a confocal laser scanning microscope (Nikon, Tokyo, Japan).
Statistics
Data are expressed as means ± SD and were analysed with one- or two-way ANOVA as appropriate, with post hoc Dunnett’s multiple comparisons test (Prism 4 software; Graphpad Software, La Jolla, CA, USA). To denote statistical significance, we have used asterisks to define differences within LDL categories (i.e. the effect of different treatments on pericyte responses to either N-LDL or HOG-LDL), and daggers to define differences between LDL categories (i.e. between N-LDL and HOG-LDL).
Results
Individually, Poly-I, NAC, 4-PBA and CsA mitigate effects of HOG-LDL on HRCP viability
HOG-LDL (200 mg/l), but not N-LDL (200 mg/l), decreased HRCP viability after 24 h incubation, consistent with previous studies [10]. This effect of HOG-LDL was partially mitigated by each of the inhibitors (Poly-I, NAC, 4-PBA and CsA (Fig. 1)), implicating oxidative stress, ER stress and mitochondrial dysfunction in HOG-LDL-induced toxicity towards HRCP. To show that the effects of NAC, 4-PBA and Poly-I could be attributed to inhibition of oxidative stress, ER stress, and scavenger receptors respectively, we confirmed our findings using alternative agents (Tempol, tauroursodeoxycholic acid and polyinosinic:polycytidylic acid [Poly-I:C]) (data not shown).

Decreased HRCP viability is induced by HOG-LDL vs N-LDL and is partially mitigated by Poly-I, NAC, 4-PBA and CsA. HRCP were pre-treated (1 h) with or without Poly-I (50 μg/ml), NAC (100 μmol/l), 4-PBA (0.5 mmol/l) or CsA (2 μmol/l) for 1 h, then treated with N-LDL or HOG-LDL (200 mg/l) for 24 h. Cell viability was measured by CCK-8 assay. HOG-LDL, but not N-LDL, decreased viability. This effect was partially blocked by each of the four agents. In comparison, pretreatment with the same four agents did not alter cell viability following exposure to N-LDL. Data, as percentage of untreated control (SFM), are expressed as mean ± SD; n = 3; *p < 0.05 vs HOG-LDL control; †† p < 0.01 vs N-LDL control
HOG-LDL induces the activation of oxidative and nitrosative stress in HRCP
Intracellular ROS increased progressively after exposure to HOG-LDL for up to 6 h (p < 0.01, n = 3); but no effect was seen with N-LDL (Fig. 2a). Levels of GPX-1, SOD-2 and 3-NT (a marker of oxidative and nitrosative stress) were detected from 0 to 24 h by western blot. GPX-1, a key enzyme of the antioxidant system, was significantly reduced following exposure to HOG-LDL vs N-LDL, while SOD-2 and 3-NT were significantly elevated, peaking at 12 h (p < 0.05, n = 3) (Fig. 2b–e). HRCP were also exposed to HOG-LDL vs N-LDL for 12 h with or without 1 h of pretreatment with Poly-I, NAC, 4-PBA and CsA. The increase of SOD-2 and 3NT, and the decrease of GPX-1 that were induced by HOG-LDL were mitigated by Poly-I and NAC pretreatment, but not by 4-PBA and CsA. In contrast to HOG-LDL, N-LDL had no effect on these responses (Fig. 2f–i).

HOG-LDL induces oxidative stress and nitrosative stress in HRCP. (a) Time course of intracellular ROS levels in HRCP. After incubation in SFM for 18 h, cells were exposed to N-LDL (grey line) or HOG-LDL (black line) (200 mg/l). Significantly higher levels of ROS were produced in response to HOG-LDL vs N-LDL. Values are mean ± SD; n = 3; †† p < 0.01; ††† p < 0.001. (b) Cells were treated with N-LDL or HOG-LDL (200 mg/l) for up to 24 h and western blot experiments performed on total protein extracts. (c) Histogram of time course for levels of GPX-1, (d) SOD-2 and (e) 3-NT in HRCP after treatment as above (b) (white bars, N-LDL; black bars, HOG-LDL). Data are expressed as fold change vs 0 h N-LDL, and are mean ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL at 0 h. (f) Poly-I and NAC, but not 4-PBA or CsA, inhibit HOG-LDL-activated oxidative and nitrosative stress in HRCP. Cells were treated with N-LDL or HOG-LDL (200 mg/l) for 12 h following pre-incubation (1 h) with or without inhibitors, and protein levels as labelled were detected by western blot performed on total protein extracts. (g) The graphs show fold changes for GPX-1, (h) SOD-2 and (i) 3-NT, expressed as mean ± SD; n = 3; *p < 0.05 vs HOG-LDL control; †† p < 0.01 vs N-LDL control
HOG-LDL induces activation of ER stress in HRCP
Levels of ER stress-related molecules (GRP78, p-eIF2α, CHOP, ATF6 and sXBP-1) were measured at several time points from 0 to 24 h by western blot, cell staining and real-time quantitative PCR following exposure of cells to N- or HOG-LDL (200 mg/l). Levels of GRP78, p-eIF2α and CHOP were all increased by HOG-LDL, but not by N-LDL, the increase occurring in a time-dependent manner and peaking at 6 or 12 h (p < 0.05, n = 3) (Fig. 3a–d). Pretreatment (1 h) of cells with Poly-I, NAC or 4-PBA blocked the effects of HOG-LDL on GPR78, p-eIF2α and CHOP abundance, but CsA had no effect (Fig. 3e–h). ATF6 translocation was observed in HOG-LDL-treated cells, but not in those treated with SFM or N-LDL (Fig. 3i). The expression of CHOP and sXBP-1 mRNA was 13- and sevenfold higher, respectively, following treatment with HOG-LDL vs N-LDL (p < 0.05, n = 3); these increases were inhibited by Poly-I, NAC and 4-PBA, but not by CsA (Fig. 3j, k).

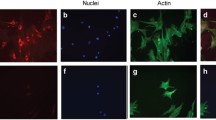
HOG-LDL activates ER stress in HRCP. (a) Time course of ER stress markers GRP78, p-eIF2α and CHOP in HRCP. Cells were treated as above (Fig. 2b). (b) Quantification of blot for GRP78, (c) p-eIF2α and (d) CHOP, expressed as means ± SD; n = 3; *p < 0.05, **p < 0.01 and ***p < 0.001 vs HOG-LDL at 0 h. White bars, N-LDL; black bars, HOG-LDL. (e) Poly-I, NAC and 4-PBA, but not CsA, inhibit HOG-LDL-activated ER stress in HRCP. Cells were treated for 12 h as above (Fig. 2f), and protein levels detected by western blot and quantified for (f) GRP78, (g) p-eIF2α and (h) CHOP. Values are mean ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL control; †† p < 0.01 and ††† p < 0.001 vs N-LDL control. (i) Immunocytochemistry images showing nuclear translocation of ATF6 in HRCP. Cells were treated with N-LDL or HOG-LDL (200 mg/l) for 12 h. Data are representative of three separate experiments and show that HOG-LDL, but not N-LDL induced ATF6 translocation from cytoplasm to nucleus. (j, k) mRNA expression in HRCP. Real-time PCR documented mRNA expression for CHOP (j) and sXBP-1 (k) in HRCP treated for 12 h with N-LDL or HOG-LDL (200 mg/l) following pre-incubation (1 h) with or without Poly-I, NAC, 4-PBA and CsA. Relative mRNA levels were normalised to 18 s mRNA. Values are means ± SD; n = 3; **p < 0.01 vs HOG-LDL control; †† p < 0.01 vs N-LDL control
HOG-LDL induces mitochondrial dysfunction in HRCP
HOG-LDL vs N-LDL significantly decreased mitochondrial membrane potential, an effect that was blocked by CsA and partially blocked by Poly-I, NAC and 4-PBA (Fig. 4a). HOG-LDL increased CYT-C levels in a time-dependent manner (Fig. 4b), an effect inhibited by Poly-I, NAC, 4-PBA and CsA (Fig. 4c).

HOG-LDL decreases mitochondrial membrane potential. (a) Mitochondrial membrane potential detection in HRCP. Cells were treated for 12 h as above (Fig. 3j, k). Mitochondrial membrane potential analyses were performed by flow cytometry with DiOC6(3), a fluorescent dye used to measure membrane potential. The percentage of total events associated with low membrane potential corresponds to low fluorescence events, which are shown as horizontal bars (denoted ‘M1’) in the histograms. Data are representative of three independent experiments. (b) Time course of CYT-C levels in HRCP. Cells were treated as above (Fig. 2b) and western blot experiments performed on total protein extracts, with β-actin used as loading control. The quantification of blots is expressed as mean ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL at 0 h. (c) Poly-I, NAC, 4-PBA and CsA inhibit HOG-LDL-induced CYT-C upregulation in HRCP. Cells were treated for 12 h as above (Fig. 2f) and CYT-C levels detected by western blot, with quantification expressed as mean ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL control; †† p < 0.01 vs N-LDL control. White bars, N-LDL; black bars, HOG-LDL
HOG-LDL induces apoptosis in HRCP
TUNEL-positive cells were increased in cells treated with HOG-LDL vs SFM or N-LDL (Fig. 5a), consistent with previous in vitro and ex vivo findings [10]. Levels of activated caspase-3 and cleaved PARP, and the ratio of BAX:BCL-2 were significantly increased by HOG-LDL vs N-LDL, peaking at 12 h (Fig. 5b–e), with apoptosis being inhibited by pre-treatment with Poly-I, NAC, 4-PBA and CsA (Fig. 5f–i). Taken together, our data suggest that HOG-LDL-induced apoptosis is caspase-dependent and downstream of oxidative stress, ER stress and mitochondrial dysfunction.

HOG-LDL induces apoptosis in HRCP. (a) TUNEL staining in HRCP. Apoptotic cells were observed by TUNEL assay when HRCP were exposed to HOG-LDL (200 mg/l) vs N-LDL (200 mg/l) for 24 h. Apoptosis significantly increased after HOG-LDL, but not after N-LDL treatment. Images are representative of three independent experiments. (b) Time course of activated caspase-3, cleaved PARP, BAX and BCL-2 in HRCP. Cells were treated as above (Fig. 2b) and western blot experiments performed on total protein extracts, with β-actin used as loading control. (c) Quantification of findings for activated caspase-3, (d) cleaved PARP and (e) BAX and BCL-2, expressed as means ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL 0 h. White bars, N-LDL; black bars, HOG-LDL. (f) Poly-I, NAC, 4-PBA and CsA inhibit HOG-LDL-induced apoptosis in HRCP. Cells were treated for 24 h as shown and protein levels as indicated detected by western blot. (g) Quantification of findings for activated caspase-3, (h) cleaved PARP and (i) BAX:BCL-2 ratio, expressed as means ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL control; †† p < 0.01 vs N-LDL control
HOG-LDL induces autophagy in HRCP
Time-dependent increases of LC3-II, beclin-1 and ATG5, all markers of autophagy [29], were induced by HOG-LDL vs N-LDL (Fig. 6a–d). When HRCP were pretreated with Poly-I, NAC, 4-PBA and CsA, increases in LC3-II and ATG5, but not in beclin-1 were inhibited (Fig. 6e–h).

HOG-LDL induces autophagy in HRCP. (a) Time course of conversion of LC3-I to LC3-II, and of ATG5 and beclin-1 abundance in HRCP. Cells were treated as above (Fig. 2b) and western blot performed on total protein extracts, with β-actin used as loading control. (b) Quantification of findings for LC3-I to LC3-II, (c) ATG5 and (d) beclin-1, expressed as means ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL 0 h. White bars, N-LDL; black bars, HOG-LDL. (e) Poly-I, NAC, 4-PBA and CsA inhibit HOG-LDL-induced autophagy in HRCP. Cells were treated for 24 h as shown, and protein conversion and abundance as indicated detected by western blot. (f) Quantification of findings for LC3-II/LC3-I ratio, (g) ATG5 and (h) beclin-1, expressed as means ± SD; n = 3; *p < 0.05 and **p < 0.01 vs HOG-LDL control; ††p < 0.01 vs N-LDL control
4-HNE and 7-KC trigger ER stress in HRCP
To define the effects of specific lipid oxidation products on ER stress in HRCP, 4-HNE and 7-KC were used. Both decreased cell viability in a dose-dependent (ESM Fig. 1a, d) and time-dependent (ESM Fig. 1b, e) manner, and increased levels of the ER stress markers GRP78, p-eIF2α and CHOP (ESM Fig. 1c, f).
Elevated levels of ER stress markers in retina from a mouse model of diabetes and hypercholesterolaemia
In retinas from a double knockout mouse model of diabetes and hypercholesterolaemia, Grp78 and Atf6 levels were significantly increased, mainly in the ganglion cell layer and inner nuclear layer, indicating increased ER stress, compared with mouse models of diabetes only or hyperlipidaemia only, or with mice that had neither condition (ESM Fig. 2).
Detection of ApoB, GRP78 and ATF6 in human retinas
To confirm the clinical relevance of our in vitro and animal findings, we performed double staining for ApoB (an LDL marker) and ER stress markers (GRP78 or ATF6) in human retinas from diabetic and non-diabetic donors (ESM Fig. 3a, b). ApoB staining was minimal in retinas from non-diabetic donors, but present in retinas from type 2 diabetic donors with non-proliferative DR. Likewise, GRP78 (ESM Fig. 3a) and ATF6 (ESM Fig. 3b) levels were minimal or absent in retinas from non-diabetic individuals, but clearly detectable in the inner retina from donors with type 2 diabetes. These findings are consistent with the notion that HOG-LDL is implicated in increasing retinal ER stress in DR.
Discussion
We conclude that HOG-LDL (a model of extravasated, modified LDL) induced CHOP-dependent apoptotic pericyte loss via enhanced oxidative and nitrosative stress, ER stress and mitochondrial dysfunction. In support of the relevance of these findings to human DR, the concentrations of LDL used were conservative estimates of those present in vivo. Thus 200 μg protein/ml is about 25% of typical plasma levels, while in atherosclerosis, intra-plaque concentrations of ApoB and oxidised LDL are 2- and 79-fold higher, respectively, than plasma levels [30, 31]. The activation of autophagy was another important consequence of exposure of pericytes to HOG-LDL and may be mediated by the same pathways. Moreover, 4-HNE or 7-KC, components of Ox-LDL, similarly induced ER stress and decreased pericyte viability, and may serve as surrogates for Ox-LDL in cell culture work. Supporting our cell culture findings, retinal ER stress was increased in a mouse model of combined diabetes and hypercholesterolaemia, but not in models of each condition alone. Finally, increased abundance of the ER chaperone, GRP78, and the ER sensor, ATF6, in human diabetic retina was accompanied by staining for Ox-LDL. These findings are consistent with (but do not prove) our hypothesis that extravasated and modified plasma lipoproteins play an important role in the propagation of DR once blood–retina barrier (BRB) leakage is established.
BRB leakage may result from several stress factors present in diabetes: elevated, fluctuating glucose levels; alterations in pH and osmotic stress; and altered thrombotic/fibrinolytic balance. It may be amplified by vicious cycles of damage caused by extravasated lipoproteins, further compromising vascular integrity. We suggest that the retinal effects of modified LDL are analogous to those in atherosclerosis, but represent a new area of DR research.
As in atherogenesis, the quantity and quality of plasma lipoproteins may be important in initiating disease. In the circulation, glycation and oxidation of LDL is relatively slight. Even so, we have previously shown that very mildly in vitro-modified (glycated and/or oxidised) LDL, simulating that found in the plasma in diabetes, decreases the viability of (bovine) retinal capillary endothelial cells and pericytes [13]. Therefore, in diabetes, the effects of mildly modified plasma LDL, combined with those of hyperglycaemia, may play a role in initiating retinal capillary damage, but the main injurious effects of LDL are likely to follow extravasation and further modification. In the retina, in contrast to arteries, lipoproteins are normally stringently excluded by the BRB, so the consequences of leakage are amplified.
Supporting a critical role for extravasated plasma lipoproteins in the propagation of DR, in a previous study we showed that Ox-LDL (detected with an antibody against copper-oxidised LDL, similar to HOG-LDL in our current work) is present in human diabetic retinas and proportional to DR severity [19]. This is the case even before the appearance of clinical disease, consistent with an initiating role for modified LDL in DR. We have also demonstrated that HOG-LDL induced apoptosis in HRCP [13, 16], but the mechanism was not explored in detail. The present study demonstrates that decreased cell viability induced by HOG-LDL was mitigated by blockade of the scavenger receptor (using Poly-I) and by inhibition of oxidative stress (with NAC), ER stress (with 4-PBA) or mitochondrial dysfunction (with low concentrations of CsA). Our data implicate all these processes in HOG-LDL-induced pericyte loss. We confirmed these findings using alterative inhibitors of oxidative stress (Tempol) and ER stress (tauroursodeoxycholic acid). Poly-I can, as shown in a recent study [32], activate toll-like receptors (TLRs) in lymphocytes, raising the possibility of an alternative mode of action. To address this question, another more powerful and classic TLR activator, Poly-I:C [33], was used. Poly-I, but not Poly-I:C mitigated the HOG-LDL-induced effects on cell viability, but neither Poly-I nor Poly-I:C affected viability (data not shown): thus the effect of Poly-I on pericytes does not seem to involve effects on TLR.
Oxidative stress has been implicated in DR [34, 35]. Excessive ROS production causes oxidative and nitrosative stress, leading to retinal capillary cell damage and death [36]. Our study shows that HOG-LDL vs N-LDL induces oxidative stress (increased intracellular ROS and 3-NT levels) in HRCP and simultaneously reduces levels of the antioxidant enzyme, GPX-1. In contrast, SOD-2 levels initially increased, presumably as a defensive response [37], then declined. Blockade of oxidative stress with NAC prevented HOG-LDL-induced ER stress and apoptosis, suggesting that oxidative stress is a proximal event.
The ER is a sensor for cellular stresses (e.g. hyperglycaemia, dyslipoproteinaemia, hypoxia, oxidative stress), and ER stress has been implicated in DR [38–40]. In this study, HOG-LDL induced phosphorylation of eIF2α, nuclear translocation of ATF6 and increased GRP78. Moreover, mRNA levels of sXBP-1 (an important transcription factor in ER stress) and CHOP (an ER-specific pro-apoptotic factor) were increased in pericytes incubated with HOG-LDL. The increase in CHOP mRNA was paralleled by increased CHOP abundance. HOG-LDL also increased levels of the pro-apoptotic factors caspase-3 and BAX, and decreased anti-apoptotic BCL-2. These effects are consistent with data showing that CHOP induces the transcription of several pro-apoptotic genes and suppresses the transcription of BCL-2 [41]. Altogether, the data indicate that HOG-LDL activates p-eIF2α and ATF6, leading to activation of pro-apoptotic mediators (CHOP) and production of the protective chaperone, GRP78. Recent studies indicate that moderate ER stress can be overcome by the unfolded protein response, but excessive and prolonged ER stress results in apoptosis [42–44]. Our data suggest that HOG-LDL-induced ER stress (particularly CHOP activation) plays an important role in promoting pericyte loss and retinal injury in DR.
Besides the ER, mitochondria also play a critical role in the development of DR and retinal capillary cell death [22]. In HRCP, we observed HOG-LDL-induced mitochondrial dysfunction, demonstrated by decreased membrane potential and release of CYT-C. We used CsA to inhibit mitochondrial dysfunction. At low concentrations such as those we employed, CsA inhibits opening of the mitochondrial permeability transition pore and has anti-apoptotic effects [45], although at higher concentrations it may induce apoptosis [46]. Treatment with CsA inhibited CYT-C release and apoptosis, supporting a role for mitochondrial dysfunction in HOG-LDL-induced apoptosis. Interestingly, blockage of ER stress with 4-PBA not only inhibited HOG-LDL-induced apoptosis, but also mitochondrial dysfunction. HOG-LDL-induced mitochondrial dysfunction may lead to apoptosis via CYT-C leakage. In addition, HOG-LDL may induce mitochondrial dysfunction via ER stress. Thus, ER stress may trigger apoptosis through two separate pathways, CHOP and CYT-C.
An interesting aspect of the current study is that, in addition to apoptosis, HOG-LDL was also found to induce autophagy, as indicated by increased levels of LC3-II, beclin-1 and ATG5. Moreover, when oxidative stress, ER stress and mitochondrial dysfunction were individually inhibited, autophagy was attenuated. This was most clearly seen in the responses of LC3-II and ATG5, while the response of beclin-1 was less conclusive. Thus, HOG-LDL-induced autophagy in HRCP is, at least in part, downstream of oxidative stress, ER stress and mitochondrial dysfunction. Several studies have shown that autophagy is not only a reparative process by which cells remove debris, alleviate ER stress and thus survive [47, 48], but may also cause cell death if ER stress is prolonged [49, 50]. In the current study, autophagy appears to play a dual role in pericytes exposed to HOG-LDL, promoting survival under mild stress, but leading to cell death under extended stress. These effects need to be addressed in detail in future studies.
We conclude that oxidative stress, ER stress and mitochondrial dysfunction are all implicated in HOG-LDL-induced HRCP apoptosis and autophagy. To elucidate the sequence of these events, inhibitors of scavenger receptor (Poly-I), oxidative stress (NAC), ER stress (4-PBA) and mitochondrial dysfunction (CsA) were employed. Poly-I and NAC blocked all the responses mentioned above; 4-PBA blocked ER stress, mitochondrial dysfunction, apoptosis and autophagy; while CsA only blocked mitochondrial dysfunction, apoptosis and autophagy. Overall, the data suggest that the initial event may be an interaction of HOG-LDL with the scavenger receptor, subsequently triggering sequential oxidative stress, ER stress and mitochondrial dysfunction, with apoptosis and autophagy as final outcomes. Considering that each of the inhibitors only partially blocked the downstream pathways, it is possible that HOG-LDL may directly induce ER stress independently of oxidative stress and directly induce mitochondrial dysfunction independently of oxidative and ER stress. Our studies employed pharmacological inhibitors, and we recognise that the absolute specificity of these agents can never be guaranteed. Studies using gene manipulation, such as gene silencing by small interfering RNA (siRNA), are now needed to elucidate these effects in greater detail.
We tested specific lipid oxidative products to determine whether their effects on HRCP resembled those of HOG-LDL. Recent reports show that 4-HNE and 7-KC can induce ER stress and autophagy [49, 51]. In this study, we found that 4-HNE and 7-KC decreased viability, triggered phosphorylation of eIF2α and increased the production of GRP78 and CHOP. Similar responses have been observed in studies of human aortic smooth muscle cells [52]. The data show that ER stress activated by HOG-LDL in HRCP can be simulated by the specific known lipoxidation products, 4-HNE and 7-KC.
Consistent with our in vitro findings, our in vivo studies also demonstrate the presence of increased ER stress in the diabetic retina and are consistent with a contribution from modified lipoproteins. In retinas from a mouse model of diabetes combined with hypercholesterolaemia, the ER stress indicators, Grp78 and Atf6, were increased; however, the increased ER stress was not observed in models of diabetes or hypercholesterolemia alone. Our studies of human retinas also supported the clinical relevance of our in vitro and animal findings. We found that GRP78 and ATF6 are present in retinas from type 2 diabetic donors with non-proliferative DR, but absent in retinas from non-diabetic donors. We also found that enhanced staining of the ER stress markers was accompanied by evidence of increased leakage of LDL (ApoB). These findings support the notion that extravasated and modified LDL is implicated in ER stress in DR.
In conclusion, our findings provide new details concerning the toxicity of extravasated, modified LDL towards retinal capillary pericytes. We also provide evidence that ER stress is present in vivo in retinas from diabetic human donors and from a murine model of diabetes, in both cases consistent with lipoproteins serving as potential causal factors. This knowledge provides a basis for the development of future, targeted interventions for the prevention and treatment of DR.
Abbreviations
- Apo:
-
Apolipoprotein
- ATF6:
-
Activating transcription factor 6
- ATG5:
-
Autophagy-related homologue 5
- BAX:
-
B-cell lymphoma 2-associated X protein (a promoter of apoptosis)
- BCL-2:
-
B-cell lymphoma 2 protein (an apoptosis regulator protein)
- BRB:
-
Blood–retina barrier
- CHOP:
-
C/EBP-homologous protein
- CsA:
-
Cyclosporin A
- CYT-C:
-
Cytochrome c
- DiOC6(3):
-
3,3′-Dihexyloxacarbocyanine iodide
- DR:
-
Diabetic retinopathy
- ER:
-
Endoplasmic reticulum
- GPX-1:
-
Glutathione peroxidase 1
- GRP78:
-
78 kDa glucose-regulated protein
- 4-HNE:
-
4-Hydroxynonenal
- HOG-LDL:
-
Highly-oxidised glycated LDL
- HRCP:
-
Human retinal capillary pericytes
- 7-KC:
-
7-Ketocholesterol
- LC3:
-
Microtubule-associated protein 1 light chain 3
- NAC:
-
N-Acetyl cysteine
- NDRI:
-
National Disease Research Interchange
- N-LDL:
-
Native LDL
- 3-NT:
-
Protein-bound 3-nitrotyrosine
- OUHSC:
-
University of Oklahoma Health Sciences Center
- Ox-LDL:
-
Oxidised LDL
- PARP:
-
Poly ADP ribose polymerase
- 4-PBA:
-
4-Phenyl butyric acid
- PDR:
-
Proliferative diabetic retinopathy
- p-eIF2α:
-
Phospho-eukaryotic initiation factor 2α
- Poly-I:
-
Polyinosinic acid
- Poly-I:C:
-
Polyinosinic:polycytidylic acid
- ROS:
-
Reactive oxygen species
- SFM:
-
Serum-free medium
- SOD-2:
-
Superoxide dismutase 2
- sXBP-1:
-
Spliced isoform of X-box binding protein 1
- TLR:
-
Toll-like receptor
- XBP-1:
-
X-box binding protein 1 (a regulator of cell stress responses)
References
Cogan DG, Toussaint D, Kuwabara T (1961) Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol 66:366–378
Kern TS, Engerman RL (1995) Vascular lesions in diabetes are distributed non-uniformly within the retina. Exp Eye Res 60:545–549
Mizutani M, Kern TS, Lorenzi M (1996) Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest 97:2883–2890
Hammes HP, Lin J, Renner O et al (2002) Pericytes and the pathogenesis of diabetic retinopathy. Diabetes 51:3107–3112
Kim J, Son JW, Lee JA, Oh YS, Shinn SH (2004) Methylglyoxal induces apoptosis mediated by reactive oxygen species in bovine retinal pericytes. J Kor Med Sci 19:95–100
Kowluru RA, Koppolu P (2002) Diabetes-induced activation of caspase-3 in retina: effect of antioxidant therapy. Free Radic Res 36:993–999
Lyons TJ, Baynes JW, Patrick JS, Colwell JA, Lopes-Virella MF (1986) Glycosylation of low density lipoprotein in patients with type 1 (insulin-dependent) diabetes: correlations with other parameters of glycaemic control. Diabetologia 29:685–689
Klein RL, Laimins M, Lopes-Virella MF (1995) Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes 44:1093–1098
Lopes-Virella MF, Klein RL, Lyons TJ, Stevenson HC, Witztum JL (1988) Glycosylation of low-density lipoprotein enhances cholesteryl ester synthesis in human monocyte-derived macrophages. Diabetes 37:550–557
Diffley JM, Wu M, Sohn M, Song W, Hammad SM, Lyons TJ (2009) Apoptosis induction by oxidized glycated LDL in human retinal capillary pericytes is independent of activation of MAPK signaling pathways. Mol Vis 15:135–145
Kobzik L (1995) Lung macrophage uptake of unopsonized environmental particulates. Role of scavenger-type receptors. J Immunol 155:367–376
Horiuchi S, Sakamoto Y, Sakai M (2003) Scavenger receptors for oxidized and glycated proteins. Amino Acids 25:283–292
Lyons TJ, Li W, Wells-Knecht MC, Jokl R (1994) Toxicity of mildly modified low-density lipoproteins to cultured retinal capillary endothelial cells and pericytes. Diabetes 43:1090–1095
Jenkins AJ, Li W, Moller K et al (1999) Pre-enrichment of modified low density lipoproteins with alpha-tocopherol mitigates adverse effects on cultured retinal capillary cells. Curr Eye Res 19:137–145
Lyons TJ, Li W, Wojciechowski B, Wells-Knecht MC, Wells-Knecht KJ, Jenkins AJ (2000) Aminoguanidine and the effects of modified LDL on cultured retinal capillary cells. Invest Ophthalmol Vis Sci 41:1176–1180
Song W, Barth JL, Lu K et al (2005) Effects of modified low-density lipoproteins on human retinal pericyte survival. Ann N Y Acad Sci 1043:390–395
Song W, Barth JL, Yu Y et al (2005) Effects of oxidized and glycated LDL on gene expression in human retinal capillary pericytes. Invest Ophthalmol Vis Sci 46:2974–2982
Barth JL, Yu Y, Song W et al (2007) Oxidised, glycated LDL selectively influences tissue inhibitor of metalloproteinase-3 gene expression and protein production in human retinal capillary pericytes. Diabetologia 50:2200–2208
Wu M, Chen Y, Wilson K et al (2008) Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Invest Ophthalmol Vis Sci 49:2679–2685
Baynes JW, Thorpe SR (1999) Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48:1–9
Li J, Wang JJ, Yu Q, Wang M, Zhang SX (2009) Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett 583:1521–1527
Kowluru RA (2005) Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal 7:1581–1587
Kowluru RA, Abbas SN (2003) Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci 44:5327–5334
Gonzalez CD, Lee MS, Marchetti P et al (2011) The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 7:2–11
Fujitani Y, Ueno T, Watada H (2010) Autophagy in health and disease. 4. The role of pancreatic beta-cell autophagy in health and diabetes. Am J Physiol Cell Physiol 299:C1–C6
Jenkins AJ, Velarde V, Klein RL et al (2000) Native and modified LDL activate extracellular signal-regulated kinases in mesangial cells. Diabetes 49:2160–2169
Fu D, Tian L, Peng Z et al (2009) Overexpression of CHMP6 induces cellular oncosis and apoptosis in HeLa cells. Biosci Biotechnol Biochem 73:494–501
Hammad SM, Powell-Braxton L, Otvos JD, Eldridge L, Won W, Lyons TJ (2003) Lipoprotein subclass profiles of hyperlipidemic diabetic mice measured by nuclear magnetic resonance spectroscopy. Metabolism 52:916–921
Klionsky DJ, Abeliovich H, Agostinis P et al (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4:151–175
Smith EB, Staples EM (1982) Plasma protein concentrations in interstitial fluid from human aortas. Proc R Soc Lond B Biol Sci 217:59–75
Nishi K, Itabe H, Uno M et al (2002) Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol 22:1649–1654
Marshall-Clarke S, Downes JE, Haga IR et al (2007) Polyinosinic acid is a ligand for toll-like receptor 3. J Biol Chem 282:24759–24766
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Kowluru RA, Chan PS (2007) Oxidative stress and diabetic retinopathy. Exp Diabetes Res 2007:43603
Madsen-Bouterse SA, Kowluru RA (2008) Oxidative stress and diabetic retinopathy: pathophysiological mechanisms and treatment perspectives. Rev Endocr Metab Disord 9:315–327
Kowluru RA, Atasi L, Ho YS (2006) Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci 47:1594–1599
Ceaser EK, Ramachandran A, Levonen AL, Darley-Usmar VM (2003) Oxidized low-density lipoprotein and 15-deoxy-delta 12,14-PGJ2 increase mitochondrial complex I activity in endothelial cells. Am J Physiol Heart Circ Physiol 285:H2298–H2308
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Ozcan U, Yilmaz E, Ozcan L et al (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140
Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 118:3378–3389
Marciniak SJ, Yun CY, Oyadomari S et al (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18:3066–3077
Gargalovic PS, Gharavi NM, Clark MJ et al (2006) The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol 26:2490–2496
Zhang K, Kaufman RJ (2008) From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462
Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789
Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S (1998) Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation 98:1153–1157
Ciechomska I, Gabrusiewicz K, Szczepankiewicz A, B Kaminska B (2012) Endoplasmic reticulum stress triggers autophagy in malignant glioma cells undergoing cyclosporine A-induced cell death. Oncogene (in press)
Albano MG, Crozet C, d’Ivernois JF (2008) Analysis of the 2004–2007 literature on therapeutic patient education in diabetes: results and trends. Acta Diabetol 45:211–219
Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281:30299–30304
Martinet W, De Meyer GR (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res 104:304–317
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752
Martinet W, De Bie M, Schrijvers DM, De Meyer GR, Herman AG, Kockx MM (2004) 7-Ketocholesterol induces protein ubiquitination, myelin figure formation, and light chain 3 processing in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24:2296–2301
Pedruzzi E, Guichard C, Ollivier V et al (2004) NAD(P)H oxidase Nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Mol Cell Biol 24:10703–10717
Acknowledgements
The NDRI provided valued assistance in obtaining human retinal tissues.
Funding
This work was supported by the Oklahoma Center for the Advancement of Science and Technology (HR08-067) and by the COBRE Program of the National Center for Research Resources (P20 RR 024215).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
DF contributed to the conception and design, acquisition of data and analysis, interpretation of data and the drafting of the article. MW contributed to the conception and design, acquisition of data and the revision of the article. JZ, MD, SMH, SY and KW contributed to the acquisition of data and revision of the article. JC contributed to the conception and design and revision of the article. TL contributed to the conception and design, writing and revision of the article. All authors gave final approval of the version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 7 kb)
ESM Fig. 1
Lipid peroxidation products decrease viability and induce ER stress in HRCP. (a) Effect of 4-HNE on HRCP viability: dose responses after 24 h exposure (means ± S.D., n = 3; *: p < 0.05; **: p < 0.01 vs. untreated control). (b) Time course for decline in HRCP viability following exposure to 4HNE (10 μM) for 1–48 h. HRCP viability was measured by CCK-8 assay (means ± S.D., n = 3; *: p < 0.05; **: p < 0.01 vs. 0 h). (c) Time course of ER stress markers GRP78, p-eIF2α and CHOP in HRCP following exposure to 4HNE (10 μM) for 1–24 h. (d) Effect of 7-KC on HRCP viability: dose responses after 24 h exposure (means ± S.D., n = 3; *: p < 0.05; **: p < 0.01 vs. untreated control). (e) Time course for decline in HRCP viability following exposure to 7-KC (20 μM) for 1–48 h. HRCP viability was measured by CCK-8 assay (means ± S.D., n = 3; *: p < 0.05; **: p < 0.01 vs. 0 h). (f) Time course of ER stress markers GRP78, p-eIF2α and CHOP in HRCP following exposure to 7-KC (20 μM) for 1–24 h. Western blot experiments were performed on total protein extracts and β-actin expression was used as loading control. Data are expressed as fold change in densitometry vs. 0 h (means ± S.D., n = 3; *: p < 0.05; **: p < 0.01 vs. 0 h) (PDF 278 kb)
ESM Fig. 2
ER stress markers are increased in retinas from hypercholesterolaemic STZ-diabetic mice. (a) ER stress sensor Grp78 and (b) Atf 6 were detected using confocal microscopy in retinas from four groups of mice (wild-type and double knockout (DKO) for Ldlr and Apobec1, with and without STZ-diabetes). There was a clear increase in both Grp78 and Atf6 staining in DKO diabetic animals (DKO-D) vs. the other three groups. (PDF 865 kb)
ESM Fig. 3
ApoB and ER stress markers are increased in retinas from human subjects with type 2 diabetes and NPDR compared with people without these conditions. Retinal sections were stained with anti-ApoB antibody (green) and (a) anti-GRP78 antibody (red) or (b) anti-ATF6 antibody (red). All sections were stained with DAPI (blue) to visualize nuclei. Images are representative of two independent experiments. Scale bar indicates 20 μm. Magnification: ×40. (ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer). There are clear increases in GRP78 and ATF6 expression, as well as in ApoB staining in the ‘diabetes with NPDR’ retinas compared to the other two categories. (PDF 681 kb)
Rights and permissions
About this article
Cite this article
Fu, D., Wu, M., Zhang, J. et al. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 55, 3128–3140 (2012). https://doi.org/10.1007/s00125-012-2692-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2692-0