
Abstract
Aims/hypothesis
Blood–retina barrier leakage in diabetes results in extravasation of plasma lipoproteins. Intra-retinal modified LDLs have been implicated in diabetic retinopathy (DR), but their effects on retinal pigment epithelial (RPE) cells and the added effects of extravasated modified HDLs are unknown.
Methods
In human retinas from individuals with and without diabetes and DR, immunohistochemistry was used to detect ApoB, ApoA1 and endoplasmic reticulum (ER) stress markers. In cell culture, human RPE cells were treated with native LDL (N-LDL) or heavily-oxidised glycated LDL (HOG-LDL) with or without pretreatment with native HDL (N-HDL) or heavily-oxidised glycated HDL (HOG-HDL). Cell viability, oxidative stress, ER stress, apoptosis and autophagy were assessed by Cell Counting Kit-8 assay, dichlorofluorescein assay, western blotting, immunofluorescence and TUNEL assay. In separate experiments, RPE cells were treated with lipid oxidation products, 7-ketocholesterol (7-KC, 5–40 μmol/l) or 4-hydroxynonenal (4-HNE, 5–80 μmol/l), with or without pretreatment with N-HDL or HOG-HDL.
Results
ApoB, ApoA1 staining and RPE ER stress were increased in the presence of DR. HOG-LDL but not N-LDL significantly decreased RPE cell viability and increased reactive oxygen species generation, ER stress, apoptosis and autophagy. Similarly, 4-HNE and 7-KC decreased viability and induced ER stress. Pretreatment with N-HDL mitigated these effects, whereas HOG-HDL was less effective by most, but not all, measures.
Conclusions/interpretation
In DR, extravascular modified LDL may promote RPE injury through oxidative stress, ER stress, autophagy and apoptosis. N-HDL has protective effects, but HOG-HDL is less effective. Extravasation and modification of HDL may modulate the injurious effects of extravasated modified LDL on the retinal pigment epithelium.
Similar content being viewed by others

Introduction
Diabetic retinopathy (DR) is the leading cause of acquired blindness among working-age adults [1]. The main risk factors include duration of diabetes, long-term severity of hyperglycaemia and hypertension [2], but other factors, including dyslipoproteinaemia [3, 4], are also implicated.
Dyslipoproteinaemia is an established risk for atherogenesis in people with and without diabetes. The term extends beyond quantitative measures and includes ‘post-synthetic’ modifications of plasma lipoproteins (e.g. by glycation and oxidation). Such modifications may occur in plasma but predominantly occur in tissues following lipoprotein extravasation and sequestration (e.g. in the arterial intima) [5, 6]. In diabetes, modification is enhanced by elevated glucose, increased oxidative stress and prolongation of exposure resulting from enhanced crosslinking to matrix proteins [7, 8]. Modified lipoproteins are considered central to the pathogenesis of atherosclerosis, wherein their harmful effects occur predominantly at the site of disease (i.e. in tissue, not in plasma).
The retina is a highly specialised tissue and extravasation of plasma constituents, including lipoproteins, is normally prevented by the inner and outer blood–retina barriers (BRBs) [9]. In DR, BRB leakage is an early feature. We hypothesised that plasma lipoproteins play a ‘hidden’ but important role in DR, critically dependent upon the presence and extent of BRB leakage and thus not primarily determined by, or related to, dyslipoproteinaemia. In support, we demonstrated immunostaining of ApoB100 (the principal apolipoprotein of LDL) and oxidised LDL (ox-LDL) in retinas of people with diabetes, even in the absence of clinical DR, and among those with DR to a greater extent and proportional to disease severity [9]. In early DR, immunostaining for ox-LDL was initially localised near the inner BRB (i.e. in the ganglion cell layer) while in more advanced DR it permeated all layers of the retina [9]. No immunostaining for ApoB or ox-LDL was found in retinas from people without diabetes.
Epidemiological data are consistent with an indirect link between dyslipoproteinaemia and DR: there are highly significant, yet relatively weak, associations between plasma lipoprotein levels and DR severity [3, 10, 11]. The weakness of these associations has, we contend, led to a lack of appreciation of the important role lipoproteins play in propagating DR, one that is only operative after BRB leakage, but significant regardless of plasma lipoprotein concentrations.
Further support for our hypothesis comes from previous cell culture work documenting the effects of modified LDL on retinal capillary endothelial cells [12] and pericytes [9, 13–16]. We showed that the mechanisms of LDL-mediated pericyte injury include oxidative and endoplasmic reticulum (ER) stresses, apoptosis and autophagy [16]. We have also demonstrated deleterious effects of modified LDL on retinal Muller cells [17], suggesting a retinal insult that extends beyond vascular cells.
Accumulating evidence suggests that early in DR, barrier leakage affects not only the inner but also the outer BRB [18]. The outer BRB is comprised of tight junctions between retinal pigment epithelial (RPE) cells. The retinal pigment epithelium is a multi-functional cell monolayer and is critically important to retinal health. Exposure of the retinal pigment epithelium to modified LDL could therefore mediate significant retinal injury, including leakage of the outer BRB.
The present study employs human tissues for immunochemistry and cell culture studies on the effects of modified LDL on human RPE cells in culture and, since extravasation of LDL implies extravasation of HDL and also since extravasated HDL is subject to the same modifying stresses as LDL, we also studied the effects of normal and modified HDL. HDL has particular relevance to the retinal pigment epithelium. There is extensive cholesterol transport across the RPE monolayer and intra-retinal ‘HDL-like particles’ mediate export of cholesterol from retinal rods and cones to the retinal pigment epithelium [19]. Two important products of lipid peroxidation, 7-ketocholesterol (7-KC) and 4-hydroxynonenal (4-HNE), were also employed to confirm findings and to define the utility of these compounds as surrogates for modified LDL in future studies.
Methods
Ethics
The study was approved by the Institutional Review Board at the University of Oklahoma Health Sciences Center.
Human retinal tissues
Normal human eyes and eyes from individuals with diabetes, fixed in 10% neutral buffered formalin within 12 h of death, were obtained from the National Diseases Research Interchange (NDRI; Philadelphia, PA, USA). Three groups (eyes from three individuals per group) were included in the study: without diabetes, with type 2 diabetes but without retinopathy and type 2 diabetes with retinopathy. Ophthalmological records provided DR status and the absence of other retinal diseases, including macular oedema. Eyes from individuals with diabetic neuropathy or nephropathy were excluded. Paraffin-embedded retinal sections (5 μm) were prepared and used for immunohistochemistry studies.
Human lipoproteins: preparation, modification and characterisation of LDL and HDL
Native LDL (N-LDL) and modified LDL were prepared as previously described [9, 20], and native HDL (N-HDL) and modified HDL were prepared using similar protocols. Briefly, human LDL and HDL were isolated from pooled plasma obtained from male and female healthy volunteers (n = 4 for each preparation), aged 20–40 years, who were not taking antioxidant vitamins or any prescribed medications. N-LDL and N-HDL were isolated by sequential ultracentrifugation (350,000 g, N-LDL density [d] = 1.019–1.063, N-HDL density [d] = 1.063–1.21) of pooled plasma. ‘Highly oxidised glycated’ (HOG-) LDL and HOG-HDL were prepared by first glycating N-LDL or N-HDL in the presence of freshly prepared 50 mmol/l d-glucose for 72 h at 37°C under antioxidant conditions (1 mmol/l diethylenetriaminepentaacetic acid (DTPA), 270 μmol/l EDTA, under nitrogen). The glycated lipoproteins were oxidised by incubation in 10 μmol/l CuCl2 (24 h, 37°C), followed by extensive dialyses to remove copper ions and glucose [20]. Protein content was determined (BCA protein assay; Pierce, Rockford, IL, USA). Lipoproteins were characterised by gel electrophoresis (Paragon Lipo Gel; Beckman, Fullerton, CA, USA), by fluorescence (360 nm excitation/430 nm emission; Gilford Fluorometer IV; Oberlin, OH, USA) and by absorbance at 234 nm (DU650 spectrophotometer; Beckman). The protocols aimed to simulate the initial in vivo glycation of lipoproteins in plasma and their subsequent oxidation after extravasation and sequestration in tissues. Lipoprotein preparations were stored in the dark under nitrogen at 4°C, and were used within 1 month of preparation. Experiments were repeated using different LDL preparations.
Human RPE cell cultures
Telomerase-immortalised human RPE (hTERT-RPE) cells (ATCC, Manassas, VA, USA) were cultured in DMEM containing 4.5 g/l glucose, 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were grown to 80–90% confluence and were made quiescent by overnight exposure to serum-free medium (SFM) before the addition of lipoproteins.
Immunohistochemistry in human diabetic retinas
Immunostaining of ApoB, ApoA1, ER stress marker 78 kDa glucose-regulated protein (GRP78; Lys-Asp-Glu-Leu) and retinal pigment epithelium-specific 65 kDa protein (RPE65) in human retinas was performed as described [9]. Briefly, retinal sections were incubated with primary anti-ApoB (1:100), anti-ApoA1 (1:100) and anti-GRP78 (1:50) (Abcam, Cambridge, MA, USA) or anti-RPE65 (1:100; Millipore, Billerica, MA, USA) (4°C, overnight), then incubated with secondary antibodies (37°C, 2 h). Immunostaining for ox-LDL and 4-HNE employed primary antibodies from Abcam. Fluorescence signals were visualised under a fluorescence microscope (Nikon E800 Epifluorescence Microscope, Tokyo, Japan).
Cell viability assay
hTERT-RPE cells were cultured in 96-well plates (15,000 cells/well). Cells were exposed to N- or HOG-LDL (200 μg protein/ml), 7-KC (5–40 μmol/l) or 4-HNE (5–80 μmol/l), with or without pretreatment with N- or HOG-HDL (500 μg/ml). Cell viability was measured by Cell Counting Kit-8 assay (CCK-8; Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer’s instructions. Briefly, treated cells were washed with SFM, incubated with CCK-8 solution (2 h, 37°C) and absorbance was measured at 450 nm.
Immunocytochemistry: activating transcription factor 6 translocation and TUNEL assay
RPE cells were cultured to 80% confluence on glass coverslips, then treated with N- or HOG-LDL (200 μg/ml) for 12 h, with or without pretreatment with N- or HOG-HDL (500 μg/ml). After fixation (4% paraformaldehyde fixation buffer, 15 min) and permeabilisation (0.5% Triton X-100 in PBS, 15 min), cells were incubated with anti-ATF6 (Abcam) (overnight, 4°C), then with Alexa Fluor 594-conjugated anti-rabbit IgG (2 h, room temperature). Fluorescence was visualised using a Nikon E800 Epifluorescence Microscope.
Cell death was detected by TUNEL assay (In Situ Cell Death Detection Kit; Roche Diagnostics, Indianapolis, IN, USA). Briefly, RPE cells grown on glass coverslips were incubated with TUNEL reaction mixture, counterstained with DAPI and positive nuclei were identified (bright green signal).
Dichlorofluorescein assay
Intracellular reactive oxygen species (ROS) were measured using a dichlorofluorescein (DCF) kit (Invitrogen, Carlsbad, CA, USA). RPE cells were studied following exposure to N- or HOG-LDL (200 μg/ml) for up to 1 h, with or without pretreatment with N- or HOG-HDL (500 μg/ml, 1 h). Briefly, cells were incubated with 10 μmol/l 2',7'-dichlorodihydrofluorescein diacetate solution (37°C, 30 min) before N-LDL treatment. DCF fluorescence was detected by a microplate reader (excitation: 485 nm; emission: 538 nm) (VICTOR3 V Multilabel Counter; PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) and expressed as a ratio of baseline levels.
Western blotting
RPE cells were treated with N- or HOG-LDL (200 μg/ml), 7-KC (10 μmol/l) or 4-HNE (20 μmol/l) for 12 h, with or without pretreatment with N- or HOG-HDL (500 μg/ml, 1 h). Cells were washed with PBS, lysed (RIPA lysis buffer; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and protein extracts (30 μg) were run on 12% SDS-PAGE gels, then transferred onto nitrocellulose filter membranes (Pall Life Sciences, Port Washington, NY, USA). Membranes were incubated with specific primary antibodies and detected with horseradish peroxidase-conjugated secondary antibodies. Probed proteins were visualised (Super Signal ELISA Femto Maximum Sensitivity Substrate; Thermo Scientific, Rockford, IL, USA) and detected (BioSpectrum Imaging System; UVP, Upland, CA, USA). Band intensities were normalised to β-actin and quantified using UVP analysing software (VisionWorksLS Image Acquisition and Analysis Software, UVP).
Data analysis
Data are presented as mean ± SE of at least three independent experiments. Differences between groups were examined using one-way or two-way ANOVA followed by Dunnett’s test. A p value of <0.05 was considered statistically significant.
Results
Detection of ApoB and ApoA1 in retinas from humans with and without diabetes and DR
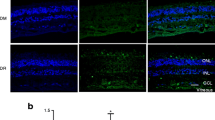
To determine whether extravasated LDL is present in the vicinity of the retinal pigment endothelium, we performed double-staining for ApoB and RPE65 in retinal sections from individuals with type 2 diabetes with and without DR, and from non-diabetic individuals (Fig. 1a). To detect HDL, we performed immunostaining for ApoA1, the principal apolipoprotein of HDL (Fig. 1b). Consistent with our previous reports [9], ApoB staining was negligible in non-diabetic retinas but was present in diabetic retinas, especially in the presence of DR. Furthermore, in retinas from individuals with DR, increased ApoB staining was observed immediately adjacent to the retinal pigment epithelium most clearly on its apical aspect, but also on the basolateral side (Fig. 1a). The staining of ApoA1 (Fig. 1b) was almost entirely in the vicinity of the retinal pigment epithelium in non-diabetic retinas, but in diabetic eyes, it was observed in the inner retina and in the presence of DR, there was widespread immunostaining throughout the retina.

Presence of ApoB and ApoA1 in the human retina. Representative retinal images for (a) ApoB (green) and RPE65 (red) and (b) ApoA1 staining (red) from five non-diabetic individuals, three individuals with type 2 diabetes with no DR and three individuals with type 2 diabetes with non-proliferative DR (NPDR). ApoA1 was detected in RPE layers in all three groups. It was also present in the inner retina of diabetic individuals and was widely distributed in those with NPDR. Magnification: (a) ×40, (b) ×20; scale bar, 100 μm. INL, inner nuclear layer; GCL, ganglion cell layer; RPE, retinal pigment epithelium; ONL, outer nuclear layer
HOG-LDL decreases RPE cell viability: protection by N-HDL but not HOG-HDL
Exposure of RPE cells to HOG-LDL (50, 100, 200, 300 μg/ml) reduced cell viability compared with SFM or N-LDL (200 μg/ml) (Fig. 2a). In time course studies, HOG-LDL (200 μg/ml) reduced RPE cell viability by 12 h (Fig. 2b). Pretreatment of cells with N-HDL (Fig. 2c) but not HOG-HDL (Fig. 2d) for 1 h prevented this effect. The protective effect of N-HDL was dose-dependent, whereas pretreatment with HOG-HDL amplified the toxic effect of HOG-LDL. HOG-HDL alone did not decrease viability. The effects of LDL and HDL on viability were visualised by microscopy (Fig. 2e–h).

HOG-LDL reduced RPE cell viability: protection by N-HDL but not HOG-HDL. RPE cells were cultured in 96-well plates to 80% confluence then made quiescent by exposure to SFM overnight. (a) Dose-dependent changes of viability in RPE cells treated with HOG-LDL for 24 h. (b) Time course of 200 μg/ml HOG-LDL-induced RPE cell viability loss. RPE cells were pretreated with (c) N-HDL or (d) HOG-HDL (both 500 μg/ml) for 1 h, then treated with 200 μg/ml HOG-LDL for 24 h. Cell viability was determined by CCK-8 assay. Data are presented as mean ± SE (*p < 0.05 vs SFM; † p < 0.05 vs HOG-LDL). Cell density changes of RPE cells treated with (e) N-LDL or (f) HOG-LDL (both 200 μg/ml) for 24 h, with and without pretreatment with (g) N-HDL or (h) HOG-HDL (both 500 μg/ml) for 1 h. Images were obtained using phase-contrast microscope. Data are representative of three independent experiments. Magnification: ×10; scale bar, 60 μm
HOG-LDL induces RPE cell apoptosis: inhibition by N-HDL but not HOG-HDL
Cells were exposed to N-LDL or HOG-LDL (200 μg/ml, 12 h), with or without pretreatment with N-HDL or HOG-HDL (500 μg/ml, 1 h). Apoptosis, detected by TUNEL assay, was markedly increased by HOG-LDL vs N-LDL, and was mitigated by pretreatment with N-HDL but not HOG-HDL (Fig. 3a). Expression of cytochrome c (Cyt-C) was markedly increased by HOG- LDL vs N-LDL, and this increase was ameliorated by N-HDL pretreatment, but not by HOG-HDL (Fig. 3b). In contrast, cleaved poly-(ADP ribose)-polymerase (PARP) was only slightly affected by the lipoproteins (Fig. 3c).

HOG-LDL induced apoptosis in RPE cells: protection by N-HDL but not HOG-HDL. Confluent RPE cells were exposed to N-LDL or HOG-LDL (both 200 μg/ml) for 12 h, with and without pre-incubation with N-HDL or HOG-HDL (both 500 μg/ml) for 1 h. (a) Representative images of TUNEL staining. Apoptotic RPE cells were revealed by TUNEL-positive labelling (green) and nuclei were counterstained with DAPI (magnification: ×40; scale bar, 20 μm). (b, c) Cell lysates were assessed for apoptotic markers Cyt-C and cleaved-PARP by western blot analysis; β-actin was loaded as protein control. Data are presented as mean ± SE (*p < 0.05 vs SFM; † p < 0.05 vs HOG-LDL)
HOG-LDL increases intracellular ROS and decreases glutathione peroxidase 1 expression in RPE cells: inhibition by N-HDL but not HOG-HDL
Treatment of RPE cells for up to 60 min with HOG-LDL vs N-LDL significantly increased the production of ROS (Fig. 4a, b) and decreased the expression of glutathione peroxidase 1 (GPX-1) (Fig. 4c, d). Pretreatment with N-HDL dramatically reduced the ROS ratio in HOG-LDL-treated cells, and HOG-HDL was less effective (Fig. 4b). In the presence of HOG-LDL, N-HDL significantly increased GPX-1 expression whereas HOG-HDL had no effect (Fig. 4d). Superoxide dismutase 2 (SOD-2) expression (western blots) was unaffected by the lipoproteins (data not shown).

HOG-LDL induced ROS production and reduced antioxidant enzyme GPX-1 expression in RPE cells: prevention by N-HDL but not HOG-HDL. ROS were measured by DCF fluorescence. Results were expressed as ratio of baseline SFM condition. (a) Confluent RPE cells were exposed to N-LDL or HOG-LDL (both 200 μg/ml) for 10, 30 and 60 min (*p < 0.05 vs SFM). Triangle, HOG-LDL; square, SFM; circle, N-LDL. (b) RPE cells were pre-incubated with N-HDL or HOG-HDL (both 500 μg/ml) for 1 h, followed by co-incubation with HOG-LDL (200 μg/ml) for 1 h (*p < 0.05 vs SFM; † p < 0.05 vs HOG-LDL). H2O2 (1 mmol/l) was used as positive control. (c) HOG-LDL (200 μg/ml) reduced GPX-1 expression in RPE over 24 h. (d) Using concentrations of LDL and HDL as in (b), the HOG-LDL-induced reduction of GPX-1 expression was mitigated by N- but not by HOG-HDL. Data are presented as mean ± SE (*p < 0.05 vs SFM; † p < 0.05 vs HOG-LDL)
HOG-LDL induces ER stress: inhibition by N-HDL but not HOG-HDL
ER stress was determined by expression of GRP78, CCAAT/enhancer-binding protein homologous protein (CHOP), phosphorylation of eIF2α and nuclear translocation of activating transcription factor 6 (ATF6). Representative western blots are shown in Fig. 5a, b and data from triplicate experiments in Fig. 5c–h. HOG-LDL vs N-LDL (200 μg/ml) increased GRP78 and CHOP expression and eIF2α phosphorylation (Fig. 5a, c–e). Pretreatment with N-HDL blocked HOG-LDL-induced ER stress, but HOG-HDL only partially inhibited CHOP expression and eIF2α phosphorylation (Fig. 5b, f–h). Translocation of ATF6 (cytoplasm to nucleus) was induced by HOG-LDL but not N-LDL at 12 h, and was prevented by pretreatment with N-HDL but not HOG-HDL (Fig. 5i).

ER stress was involved in HOG-LDL-induced RPE cell injury: protection by N-HDL and partial protection by HOG-HDL. (a, b) Representative western blots performed on total protein extracts are shown. (a) Time course of ER stress protein expression in RPE cells treated with N-LDL or HOG-LDL (200 μg/ml). (b) RPE cells were pretreated with N-HDL or HOG-HDL (500 μg/ml) for 1 h, followed by co-incubation with HOG-LDL (200 μg/ml) for 12 h. (c–e) Quantification of blots from triplicate experiments as in (a). (f–h) Quantification of blots from triplicate experiments as in (b). (i) Representative images of cytoplasmic staining and nuclear translocation of ATF6 in RPE cells treated with N-LDL or HOG-LDL (200 μg/ml) for 12 h, with and without pre-incubation with N-HDL or HOG-HDL (500 μg/ml) for 1 h (magnification: ×40; scale bar, 20 μm)
HOG-LDL induces autophagy: inhibition by both N-HDL and HOG-HDL
During autophagosome formation, conversion of unconjugated soluble microtubule-associated protein 1 light chain 3 (LC3)-I to its conjugated form, LC3-II, is a marker of autophagy. HOG-LDL vs N-LDL (200 μg/ml, 12–24 h) increased LC3-II expression (Fig. 6a). Pretreatment with either N-HDL or HOG-HDL (500 μg/ml, 1 h) inhibited HOG-LDL-induced LC3-II expression (Fig. 6b). Expression of another autophagy-related protein, Beclin-1, was not altered by HOG-LDL or N-LDL, or by pretreatment with N-HDL or HOG-HDL (western blots in Fig. 6a, b).

HOG-LDL increased autophagy in RPE cells: mitigation by N- and HOG-HDL. (a) Time course of LC3-II expression in RPE cells treated with N-LDL or HOG-LDL (200 μg/ml). (b) RPE cells were pretreated with N-HDL or HOG-HDL (500 μg/ml) for 1 h, followed by HOG-LDL (200 μg/ml) treatment for 12 h. Western analysis were performed on total protein extracts and blots were quantified relative to control (*p < 0.05 vs SFM; † p < 0.05 vs HOG-LDL)
GRP78 expression is increased in retinal pigment epithelium of retinas from individuals with DR vs non-DR diabetic or non-diabetic individuals
To define the clinical relevance of our in vitro findings, we performed double-staining for GRP78 and RPE65 in retinas from diabetic individuals with and without DR and from non-diabetic individuals (Fig. 7). GRP78 staining was very faint and homogenous in retinas from non-diabetic individuals and from diabetic individuals without DR, but was increased in diabetic retinas in the presence of DR (Fig. 7). Merged images (yellow) revealed partial co-localisation of GRP78 in the retinal pigment epithelium layer, suggesting a role for ER stress in RPE cells in the pathogenesis of DR.

Detection of GRP78 in retinal sections from non-diabetic human subjects, type 2 diabetes with and without diabetic retinopathy. Human retinal sections were stained with antibody against GRP78 (green) and RPE65 (red), and the nuclei were counterstained with DAPI (blue). Images were representative of three independent experiments. Yellow in merged images represents overlapping of green and red signals, indicating increased expression of GRP78 in human RPE layer. Magnification: ×60; scale bar, 20 μm. INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium
7-KC and 4-HNE decreased RPE cell viability and induced ER stress: protection by N-HDL and partial protection by HOG-HDL
LDL is a large particle and during oxidation numerous products are generated, including 7-KC and 4-HNE. We treated RPE cells with these specific lipid oxidation products and the results are shown in electronic supplementary materials (ESM) Figs 1, 2. Both 7-KC and 4-HNE decreased RPE cell viability in a dose-dependent manner (ESM Fig. 1) and increased levels of the ER stress markers p-eIF2α, X-box binding protein-1 and CHOP (ESM Fig. 2). The effects of 7-KC and 4-HNE were effectively mitigated by pretreatment with N-HDL but less so by HOG-HDL.
Detection of ox-LDL and 4-HNE in human retinas
We observed increased staining for ox-LDL and 4-HNE in diabetic retinas in the vicinity of the retinal pigment epithelium, more marked in the presence of clinical DR (ESM Fig. 3).
Discussion
The present study provides further support for our hypothesis that plasma lipoproteins play a central role in the propagation of DR, but one that is mainly operative after BRB leakage has led to lipoprotein extravasation and modification. We contend that modified lipoproteins then mediate toxicity towards many different retinal cell types, both vascular and non-vascular. In support of this, we previously identified the presence of extensive amounts of modified LDL in the diabetic retina and we defined its toxicity towards retinal capillary pericytes [9, 13–15], endothelial cells [12] and Muller cells [17].
This study is the first to address the effects of modified LDL on the retinal pigment epithelium, and to investigate the role of extravasated modified HDL. In human diabetic retinas, we demonstrate increased presence of ApoB adjacent to the retinal pigment epithelium on both sides, augmenting previous findings [9]. Supporting an involvement of HDL, we demonstrate increased ApoA1 staining in diabetic retinas, consistent with HDL extravasation. Such staining in diabetic retina was previously demonstrated by Sima et al [21] and was accompanied by increased expression of mRNA. This was attributed to increased production by the retinal pigment epithelium and we contend that HDL leakage is also likely to contribute. Furthermore, we demonstrate the presence of ox-LDL and 4-HNE in the vicinity of the retinal pigment epithelium in DR, using immunohistochemistry (ESM Fig. 3). We also demonstrate RPE ER stress in the presence of DR, consistent with effects of modified LDL in our cell culture studies.
In cell culture, we show that modified LDL is toxic towards RPE cells, consistent with mediation of outer BRB injury and with recent evidence that not only the inner but also the outer BRB is compromised in DR [18]. We define mechanisms for the effects of modified LDL on the retinal pigment epithelium, and show that native HDL is protective, whereas modified HDL is, in general, not protective.
Our overall hypothesis draws analogies with the role of ox-LDL in atherosclerosis, but with important differences. First, in arteries, LDL extravasation begins in childhood [22] whereas in healthy retina, lipoprotein extravasation is rigorously prevented throughout life. Thus, with both BRBs intact, dyslipoproteinaemia may be largely irrelevant to retinal function, but once BRB breakdown occurs, as in diabetes, the ‘fold increase’ in extravasated lipoproteins between diabetic vs healthy retina may greatly exceed that in arterial intima. Second, the retina differs markedly in structure from the artery. The retinal ‘neuropile’ is embryologically part of the brain [23] and although only approximately 0.5 mm thick, comprises a complex assembly of specialised cell types with negligible interstitial space: both BRBs are immediately adjacent to neurons and neural support cells. The retina consumes more oxygen per gram [24] and has a higher blood flow [25, 26] than any other tissue; it also contains a high concentration of unsaturated, oxidisable lipids. In DR, the protection normally provided by the intact BRB is compromised, exposing many different retinal cell types to modified, cytotoxic plasma lipoproteins.
Leakage of the inner BRB is well-recognised in DR and recent work by ourselves [9] and others [18] suggests that the outer BRB is also compromised. The outer BRB comprises tight junctions between cells of the RPE monolayer, and separates the choroidal circulation from the neural retina. The retinal pigment epithelium performs numerous other functions critical to intra-retinal homeostasis [27, 28], including control of cholesterol flux [29, 30]. We now show that HOG-LDL but not N-LDL causes apoptosis of RPE cells, and that this effect is mediated by oxidative stress, ER stress and autophagy. The injurious effects of HOG-LDL were ameliorated by pretreatment with native HDL; pretreatment with HOG-HDL was less effective and sometimes enhanced toxicity (e.g. decreased cell viability compared with HOG-LDL alone). As in previous work with retinal capillary pericytes and Muller cells [9, 13, 15–17], we endeavoured to ensure that cell culture conditions (LDL concentration, extent of modification) simulated stresses present in vivo in human DR.
We demonstrate that in RPE cells, HOG-LDL induces ROS production and decreases levels of GPX-1. Increased oxidative stress may damage susceptible lipids, proteins and other biomolecules and is a recognised mediator of atherosclerosis [31, 32], diabetes [33] and (as found in our own recent work) pericyte injury in DR [16, 17]. GPX-1 is the most important cytosolic and mitochondrial antioxidant enzyme in humans. It detoxifies hydrogen peroxide and lipid peroxides and its decreased activity is associated with increased risk for cardiovascular disease and diabetes [34, 35]. HDL has known antioxidant effects and in the present study of RPE cells, N-HDL completely suppressed the HOG-LDL-induced increase in ROS, but HOG-HDL was much less effective.
The ER serves vital functions, including lipid and protein synthesis, maturation and transportation, and ‘ER stress’ is linked to major diseases including cancer, neurodegenerative diseases and diabetes [36]. ER stress, mediated by excessive fatty acid flux, may induce protein misfolding and trigger the ‘unfolded protein response’ [37]. In the diabetic eye, ER stress has been implicated in the death of retinal neurons and vascular cells [38]. The ER is sensitive to oxidative stress induced by exposure to ox-LDL, as shown in our studies of retinal pericytes [16, 17]. In the present study, ER stress was induced by HOG-LDL in human RPE cells, and this effect was inhibited by pretreatment with N-HDL but only partially by HOG-HDL. Prolonged ER stress ultimately results in cell apoptosis via activation of the pro-apoptotic transcription factor CHOP and in our study HOG-LDL dramatically induced CHOP expression. Both N- and HOG-HDL inhibited the increase in CHOP expression.
Autophagy plays a key role in maintaining the intracellular environment, removing damaged organelles, cell membranes and proteins, and preserving nutrients for energy [39]. It may be induced by ER stress, which in turn it may alleviate by promoting degradation of damaged proteins [40]. Prolonged ER stress may switch autophagy from a protective role to one promoting cell death. Recent evidence implicates autophagy in the intracellular degradation of ox-LDL in human vascular endothelial cells [41]. We have shown that autophagy mediates the effects of HOG-LDL on retinal pericytes and Muller cells [16, 17]. In the present work, we show that treatment of RPE cells with HOG-LDL promotes conversion of LC3 from its cytosolic (LC3-І) to its membrane-bound form (LC3-II), an essential step in autophagosome formation. Not only N-HDL but also HOG-HDL prevents this effect. The mechanisms whereby both N-HDL and HOG-HDL can block CHOP activation and prevent the activation of autophagy need further study, and this emphasises the great complexity and multiple functions of HDL [42].
It is reported that in atherosclerosis ox-LDL triggers Cyt-C release, leading to caspase activation and vascular cell apoptosis, thereby promoting plaque formation [43]. We have documented HOG-LDL-induced apoptosis in retinal pericytes and Muller cells [9, 16, 17], and our present results with RPE cells are analogous: HOG-LDL induces apoptosis through upregulation of pro-apoptotic Cyt-C and PARP. N-HDL protects RPE cells from HOG-LDL-induced apoptosis by blocking expression of these proteins, but HOG-HDL is ineffective.
HDL is generally considered to be atheroprotective by removing excess cholesterol from cells, inhibiting lipid oxidation and inhibiting inflammatory responses [44–46]. Qualitative changes in HDL occur in diabetes, including enhanced glycation of ApoA1 molecules [47], and these may impair HDL function. For example, the capacity of HDL for reverse cholesterol transport is reduced by glycation [48] or oxidation [49]. Consistent with this, the anti-atherogenic properties of HDL isolated from the plasma of individuals with diabetes are impaired compared with HDL isolated from non-diabetic individuals [50]. This implies that once BRB leakage occurs, the diabetic retina suffers a double insult from both LDL and HDL: not only do the lipoproteins gain access to a space from which they are normally excluded but also the quality of those lipoproteins is already compromised by glycation in plasma, and is further compromised by subsequent modification.
Finally, we tested the effects of two model compounds of lipoprotein oxidation, 7-KC and 4-HNE. These are much more readily studied than modified LDL and had effects on RPE cells that were generally similar to those of HOG-LDL. They may serve as useful surrogates for HOG-LDL, at least for exploratory experiments.
In conclusion, we demonstrate that HOG-LDL may cause oxidative stress, ER stress and autophagy in RPE cells, thus mediating RPE cell apoptosis, and potentially contributing to compromise of the outer BRB. In DR, immunostaining for ApoB shows its presence in the vicinity of the retinal pigment epithelium, and for ApoA1 suggests extravasation of HDL. N-HDL completely blocked the oxidative stress, ER stress, autophagy and apoptosis induced by HOG-LDL. Modification of HDL, first by glycation then by oxidation, abolished some but not all its protective properties, and by one measure rendered it actively toxic, emphasising the structural and metabolic complexity of this particle. Overall, the findings suggest an important new role for extravasated and modified plasma lipoproteins in promoting DR: they may compromise the many important functions of the retinal pigment epithelium, including the integrity of the outer BRB. Future studies should define the differential effects of modified LDL on the apical and baso-lateral aspects of the retinal pigment epithelium. Improved understanding of these disease mechanisms may lead to new strategies to treat and prevent the development and progression of DR.
Abbreviations
- ATF6:
-
Activating transcription factor 6
- BRB:
-
Blood–retina barrier
- CCK-8:
-
Cell Counting Kit-8
- CHOP:
-
C/EBP-homologous protein
- Cyt-C:
-
Cytochrome c
- DCF:
-
Dichlorofluorescein
- DR:
-
Diabetic retinopathy
- ER:
-
Endoplasmic reticulum
- GPX-1:
-
Glutathione peroxidase 1
- GRP78:
-
78 kDa glucose-regulated protein
- 4-HNE:
-
4-Hydroxynonenal
- HOG-HDL:
-
Highly oxidised glycated HDL
- HOG-LDL:
-
Highly oxidised glycated LDL
- hTERT-RPE:
-
Telomerase-immortalised human RPE
- 7-KC:
-
7-Ketocholesterol
- LC3:
-
Microtubule-associated protein 1 light chain 3
- NDRI:
-
National Disease Research Interchange
- N-HDL:
-
Native HDL
- N-LDL:
-
Native LDL
- Ox-LDL:
-
Oxidised LDL
- PARP:
-
Poly ADP ribose polymerase
- ROS:
-
Reactive oxygen species
- RPE:
-
Retinal pigment epithelial
- RPE65:
-
Retinal pigment epithelium-specific 65 kDa protein
- SFM:
-
Serum-free medium
- SOD-2:
-
Superoxide dismutase 2
References
Fong DS, Aiello LP, Ferris FL 3rd, Klein R (2004) Diabetic retinopathy. Diabetes Care 27:2540–2553
West KM, Erdreich LJ, Stober JA (1980) A detailed study of risk factors for retinopathy and nephropathy in diabetes. Diabetes 29:501–508
Lyons TJ, Jenkins AJ, Zheng D et al (2004) Diabetic retinopathy and serum lipoprotein subclasses in the DCCT/EDIC cohort. Investig Ophthalmol Vis Sci 45:910–918
Klein R, Sharrett AR, Klein BE et al (2002) The association of atherosclerosis, vascular risk factors, and retinopathy in adults with diabetes : the atherosclerosis risk in communities study. Ophthalmology 109:1225–1234
Matsuura E, Kobayashi K, Tabuchi M, Lopez LR (2006) Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog Lipid Res 45:466–486
Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL (1989) Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med 320:915–924
Lyons TJ, Baynes JW, Patrick JS, Colwell JA, Lopes-Virella MF (1986) Glycosylation of low density lipoprotein in patients with type 1 (insulin-dependent) diabetes: correlations with other parameters of glycaemic control. Diabetologia 29:685–689
Ravandi A, Kuksis A, Shaikh NA (2000) Glucosylated glycerophosphoethanolamines are the major LDL glycation products and increase LDL susceptibility to oxidation: evidence of their presence in atherosclerotic lesions. Arterioscler Thromb Vasc Biol 20:467–477
Wu M, Chen Y, Wilson K et al (2008) Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Investig Ophthalmol Vis Sci 49:2679–2685
van Leiden HA, Dekker JM, Moll AC et al (2002) Blood pressure, lipids, and obesity are associated with retinopathy: the hoorn study. Diabetes Care 25:1320–1325
Lloyd CE, Klein R, Maser RE, Kuller LH, Becker DJ, Orchard TJ (1995) The progression of retinopathy over 2 years: the Pittsburgh Epidemiology of Diabetes Complications (EDC) Study. J Diabetes Complications 9:140–148
Lyons TJ, Li W, Wells-Knecht MC, Jokl R (1994) Toxicity of mildly modified low-density lipoproteins to cultured retinal capillary endothelial cells and pericytes. Diabetes 43:1090–1095
Song W, Barth JL, Lu K et al (2005) Effects of modified low-density lipoproteins on human retinal pericyte survival. Ann N Y Acad Sci 1043:390–395
Song W, Barth JL, Yu Y et al (2005) Effects of oxidized and glycated LDL on gene expression in human retinal capillary pericytes. Investig Ophthalmol Vis Sci 46:2974–2982
Diffley JM, Wu M, Sohn M, Song W, Hammad SM, Lyons TJ (2009) Apoptosis induction by oxidized glycated LDL in human retinal capillary pericytes is independent of activation of MAPK signaling pathways. Mol Vis 15:135–145
Fu D, Wu M, Zhang J et al (2012) Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 55:3128–3140
Wu M, Yang S, Elliott MH et al (2012) Oxidative and endoplasmic reticulum stresses mediate apoptosis induced by modified LDL in human retinal Muller cells. Investig Ophthalmol Vis Sci 53:4595–4604
Xu HZ, Le YZ (2011) Significance of outer blood-retina barrier breakdown in diabetes and ischemia. Investig Ophthalmol Vis Sci 52:2160–2164
Tserentsoodol N, Gordiyenko NV, Pascual I, Lee JW, Fliesler SJ, Rodriguez IR (2006) Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol Vis 12:1319–1333
Jenkins AJ, Velarde V, Klein RL et al (2000) Native and modified LDL activate extracellular signal-regulated kinases in mesangial cells. Diabetes 49:2160–2169
Simo R, Garcia-Ramirez M, Higuera M, Hernandez C (2009) Apolipoprotein A1 is overexpressed in the retina of diabetic patients. Am J Ophthalmol 147(319–325):e311
Holman RL, Mc GH Jr, Strong JP, Geer JC (1958) The natural history of atherosclerosis: the early aortic lesions as seen in New Orleans in the middle of the of the 20th century. Am J Pathol 34:209–235
Curcio CA, Johnson M, Huang JD, Rudolf M (2010) Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J Lipid Res 51:451–467
Yu DY, Cringle SJ (2001) Oxygen distribution and consumption within the retina in vascularised and avascular retinas and in animal models of retinal disease. Prog Retin Eye Res 20:175–208
Friedman E, Kopald HH, Smith TR (1964) Retinal and choroidal blood flow determined with krypton-85 anesthetized animals. Investig Ophthalmol 3:539–547
Feke GT, Tagawa H, Deupree DM, Goger DG, Sebag J, Weiter JJ (1989) Blood flow in the normal human retina. Investig Ophthalmol Vis Sci 30:58–65
Steinberg RH (1985) Interactions between the retinal pigment epithelium and the neural retina. Doc Ophthalmol 60:327–346
Strauss O (2005) The retinal pigment epithelium in visual function. Physiol Rev 85:845–881
Gordiyenko N, Campos M, Lee JW, Fariss RN, Sztein J, Rodriguez IR (2004) RPE cells internalize low-density lipoprotein (LDL) and oxidized LDL (oxLDL) in large quantities in vitro and in vivo. Investig Ophthalmol Vis Sci 45:2822–2829
Zheng W, Reem RE, Omarova S et al (2012) Spatial distribution of the pathways of cholesterol homeostasis in human retina. PLoS One 7:e37926
White CR, Brock TA, Chang LY et al (1994) Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci U S A 91:1044–1048
Miller FJ Jr, Gutterman DD, Rios CD, Heistad DD, Davidson BL (1998) Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res 82:1298–1305
Johansen JS, Harris AK, Rychly DJ, Ergul A (2005) Oxidative stress and the use of antioxidants in diabetes: linking basic science to clinical practice. Cardiovasc Diabetol 4:5
Lewis P, Stefanovic N, Pete J et al (2007) Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 115:2178–2187
Maritim AC, Sanders RA, Watkins JB 3rd (2003) Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol 17:24–38
Lin JH, Walter P, Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3:399–425
Salminen A, Kauppinen A, Hyttinen JM, Toropainen E, Kaarniranta K (2010) Endoplasmic reticulum stress in age-related macular degeneration: trigger for neovascularization. Mol Med 16:535–542
Oshitari T, Hata N, Yamamoto S (2008) Endoplasmic reticulum stress and diabetic retinopathy. Vasc Health Risk Manag 4:115–122
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX (2008) Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ 15:422–425
Zhang YL, Cao YJ, Zhang X et al (2010) The autophagy-lysosome pathway: a novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem Biophys Res Commun 394:377–382
Heinecke JW (2012) The not-so-simple HDL story: a new era for quantifying HDL and cardiovascular risk? Nat Med 18:1346–1347
Li Y, Higashi Y, Itabe H, Song YH, Du J, Delafontaine P (2003) Insulin-like growth factor-1 receptor activation inhibits oxidized LDL-induced cytochrome C release and apoptosis via the phosphatidylinositol 3 kinase/Akt signaling pathway. Arterioscler Thromb Vasc Biol 23:2178–2184
Assmann G, Nofer JR (2003) Atheroprotective effects of high-density lipoproteins. Annu Rev Med 54:321–341
Negre-Salvayre A, Dousset N, Ferretti G, Bacchetti T, Curatola G, Salvayre R (2006) Antioxidant and cytoprotective properties of high-density lipoproteins in vascular cells. Free Radic Biol Med 41:1031–1040
Parthasarathy S, Barnett J, Fong LG (1990) High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim Biophys Acta 1044:275–283
Berthezene F (1996) Non-insulin dependent diabetes and reverse cholesterol transport. Atherosclerosis 124(Suppl):S39–S42
Nobecourt E, Davies MJ, Brown BE et al (2007) The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin:cholesterol acyltransferase. Diabetologia 50:643–653
Sharma N, Desigan B, Ghosh S, Sanyal SN, Ganguly NK, Majumdar S (1999) The role of oxidized HDL in monocyte/macrophage functions in the pathogenesis of atherosclerosis in rhesus monkeys. Scand J Clin Lab Invest 59:215–225
Kalogerakis G, Baker AM, Christov S et al (2005) Oxidative stress and high-density lipoprotein function in type I diabetes and end-stage renal disease. Clin Sci (Lond) 108:497–506
Acknowledgements
The NDRI provided valued assistance in obtaining human retinal tissues.
Funding
This work was supported by the COBRE Program of the National Center for Research Resources (P20 RR 024215), the American Diabetes Association (07-12-CT-46), the LINJO fund and by the Oklahoma Center for the Advancement of Science and Technology (HR08-067).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
MD contributed to the conception and design of the study and to acquisition of data and analysis, interpretation of data and the drafting of the paper. MW and JC contributed to the conception and design of the study and to revision of the paper. DF, SY and KW contributed to the acquisition of data and revision of the paper. TJL contributed to the conception and design of the study and to writing and revision of the paper. All authors gave approval of the final version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Lipid peroxidation products reduced RPE cell viability; protection by N-HDL and partial protection by HOG-HDL. Dose responses to (a) 7-KC and (b) 4-HNE after 24 h exposure. RPE cells were pretreated with (c) N-HDL or (d) HOG-HDL for 1 h, followed by exposure to 7-KC (10 μmol/l, 24 h). RPE cells were pretreated with (e) N-HDL or (f) HOG-HDL for 1 h, followed by exposure to 4-HNE (20 μmol/l, 24 h). Data are presented as means ± S.E. (*P < 0.05 vs. SFM; †P < 0.05 vs. 7-KC or 4-HNE). (PDF 115 kb)
ESM Fig. 2
Lipid peroxidation products induced ER stress in RPE cells; protection by N-HDL and partial protection by HOG-HDL. ER stress markers (a) p-eIF2α, (b) CHOP, and (c) X-box binding protein-1 (XBP-1) expression in RPE cells pretreated with N-HDL or HOG-HDL (both 500 μg/ml) for 1 h, following exposure to 7-KC (10 μmol/l) or 4-HNE (20 μmol/l) for 12 h. Western blot experiments were performed on total protein extracts and β-actin expression was used as loading control. Data are presented as means ± S.E. (*P < 0.05 vs. SFM; †P < 0.05 vs. 7-KC; ‡P < 0.05 vs. 4-HNE). (PDF 1230 kb)
ESM Fig. 3
Ox-LDL and 4-HNE are increased in retinas from human subjects with type 2 diabetes and NPDR compared with people without these conditions. Retinal sections were stained with (a) ox-LDL (green) and RPE65 (red), and (b) 4-HNE (green) and RPE65 (red). All sections were stained with DAPI (blue) to visualize nuclei. Images are representative of two independent experiments. Magnification: (a) 40 x, (b) 60 x; scale bar, 50 μm. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. (PDF 873 kb)
Rights and permissions
About this article
Cite this article
Du, M., Wu, M., Fu, D. et al. Effects of modified LDL and HDL on retinal pigment epithelial cells: a role in diabetic retinopathy?. Diabetologia 56, 2318–2328 (2013). https://doi.org/10.1007/s00125-013-2986-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2986-x