
Abstract
Aims/hypothesis
Recent studies indicate that an aberrant gut microbiota is associated with the development of type 1 diabetes, yet little is known about the microbiota in children who have diabetes at an early age. To this end, the microbiota of children aged 1–5 years with new-onset type 1 diabetes was compared with the microbiota of age-matched healthy controls.
Methods
A deep global analysis of the gut microbiota composition was established by phylogenetic microarray analysis using a Human Intestinal Tract Chip (HITChip).
Results
Principal component analyses highlighted the importance of age when comparing age-matched pairs. In pairs younger than 2.9 years, the combined abundance of the class Bacilli (notably streptococci) and the phylum Bacteroidetes was higher in diabetic children, whereas the combined abundance of members of Clostridium clusters IV and XIVa was higher in the healthy controls. Controls older than 2.9 years were characterised by a higher fraction of butyrate-producing species within Clostridium clusters IV and XIVa than was seen in the corresponding diabetic children or in children from the younger age groups, while the diabetic children older than 2.9 years could be differentiated by having an increased microbial diversity.
Conclusions/interpretation
The results from both age groups suggest that non-diabetic children have a more balanced microbiota in which butyrate-producing species appear to hold a pivotal position.
Similar content being viewed by others


Introduction
The role of the intestinal microbiota as an integral determinant of human health has become increasingly evident over the past decade [1–3]. The human host and its intestinal microbes have co-evolved, resulting in the development of complex immune mechanisms that confine the commensal bacteria to the intestinal lumen and protect the host from invading pathogens. The intestinal microbiota thrives mainly on diet-derived nutrients, while the host benefits from metabolites (e.g. butyrate) from the microbial fermentation of carbohydrates that are otherwise poorly digested [4].
Various studies have shown that specific (combinations of) microbial groups can induce specific immune responses [5–7]. For example, the ratio of Firmicutes to Bacteroidetes has been shown to play an important role in the development of type 1 diabetes in rats [8, 9]. Bacteroidetes have more recently also been shown to be more abundant in (pre)diabetic children [10–13]. Gut permeability appears to be an important factor in the relationship between the intestinal microbiota and the development of type 1 diabetes [14]. Recent studies suggest that the degradation and fermentation of carbohydrates into short-chain fatty acids in cross-feeding relationships between microbial groups determine the level of permeability [15, 16]. Adequate butyrate production levels in particular are predicted to be essential for gut integrity [15, 17], which is exemplified by the fact that butyrate inhibits inflammatory responses [18]. Furthermore, the butyrate-producing bacterium Faecalibacterium prausnitzii is known to have anti-inflammatory effects both in vitro and in vivo [7, 19], and has been found to be decreased in abundance in children with three or more diabetes-related autoantibodies [11].
Like many autoimmune diseases, the incidence of type 1 diabetes is increasing worldwide, showing a particularly sharp increase among children under the age of 5 years [20–23]. The aim of this study was to compare the gut microbiota of 1- to 5-year-old diabetic children with that of age-matched non-diabetic control children using the Human Intestinal Tract Chip (HITChip), a phylogenetic microarray that provides a deep global compositional analysis of the human intestinal microbiota [24].
Methods
Sample collection
Faecal samples were collected from children newly diagnosed with type 1 diabetes in different European countries during the years 2004–2010. Patients were recruited into two research projects – the DIPP (Finnish Type 1 Diabetes Prediction and Prevention) study in Finland (http://dipp.utu.fi/) and the international VirDiab (Viruses in Diabetes) study (www.uta.fi/med/virdiab/index.html), which included cases and control children from seven European countries. Altogether in this study, DNA was successfully isolated from 28 diabetic children, including four children from France, one from Greece, three from Estonia, two from Lithuania and 18 from Finland (see electronic supplementary material [ESM] Table 1). The diabetic children were matched with control children according to age; DNA was isolated successfully from 27 control children. One of the control children was from Lithuania and the rest were from Finland (ESM Table 1). The samples were collected from the diabetic children and their corresponding controls within 4 weeks of the diagnosis of diabetes. Samples were collected by the parents at home and shipped by mail at ambient temperature to the laboratory, where they were subsequently stored at −75°C.
DNA extraction
Total DNA was extracted from a 0.25 g faecal sample using the repeated bead beating method described in detail by Yu and Morrison [25], with a number of modifications. In brief, four 3 mm instead of 0.5 mm glass beads were added during the homogenisation step. Bead beating was performed using a Precellys 24 (Bertin Technologies, Montigny le Bretonneux, France) at 5.5 beats/ms in three rounds of 1 min each with 30 s pauses at room temperature in between. The incubation temperature after the bead beating was raised from 70°C to 95°C. Importantly, protein precipitation with 260 μl of ammonium acetate was carried out twice instead of only once. Elution of DNA from the purification columns was done twice. Columns from the QiaAmp Stool Kit were replaced by those from the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany).
HITChip analysis
The intestinal microbiota composition was assessed using the HITChip, a phylogenetic profiling DNA microarray containing over 5,000 probes based on the 16S rRNA gene sequences of over 1,100 intestinal bacterial phylotypes. This microarray identifies both the variation and the relative quantity of the human intestinal tract communities at Level 1 (phylum-like, with Firmicutes divided into classes or clusters), Level 2 (groups at an approximate genus level with >90% 16S rRNA identity) and/or Level 3 (phylotypes based on >98% sequence identity) [24]. Hybridisations were performed in duplicate with samples labelled with Cy3 and Cy5 dyes. The slides were scanned, and the data extracted from the microarray images using Agilent Feature Extraction software, version 10.7.3.1 (www.agilent.com). Array normalisation was performed as previously described [24, 26] using a set of R-based scripts (http://r-project.org) in combination with a custom-designed database that runs under the MySQL database management system (www.mysql.com). This was implemented on both dyes for each sample, and duplicate hybridisations with a Pearson correlation >0.98 were considered for further analysis. Complete linkage was used for the construction of hierarchical clusters of the total microbiota probe profiles.
Statistical analysis
Initial analysis of the samples showed that the bacterial populations present in the gut were most often not normally distributed. Non-parametric tests were therefore used. Case–control samples were matched based on age, and this was done before the HITChip analysis was carried out. DNA extraction failed in a few samples, resulting in three unpaired samples (ESM Table 1) whose data were only used in the unpaired analyses. All tests were two-tailed. Values of p < 0.05 were considered to indicate statistical significance. Data is reported as mean ± SD. In addition, principal component analysis (PCA) was performed to find clusters of similar groups of samples or species. PCA is an ordination method based on multivariate statistical analysis that maps the samples in different dimensions. All tests were performed with PASW Statistics 18 (SPSS, Chicago, IL, USA). Redundancy analyses were performed with Canoco 4.5 (Microcomputer Power, Ithaca, USA).
Results
The microbial composition of faecal samples from 27 healthy controls and 28 diabetic children (age 1.3–4.6 years; mean ± SD 2.7 ± 0.8; constituting 26 sample pairs matched based on the child’s age) was determined by HITChip analysis (ESM Table 1). This revealed that Clostridium clusters XIVa (56%) and IV (11%) were most abundant in the faeces of the children, followed by Actinobacteria (9.2%), Bacilli (8.5%) and Bacteriodetes (7.5%) (ESM Fig. 1). Importantly, analysing the phylogenetic differences between the diabetic and control children using PCA, redundancy analysis and diversity analysis showed that the control–diabetic pairs should be separated into two groups based on their age, with a cut-off around the age of 3 years.
PCA on the genus-like level
In the PCA plots of the genus-like level, controls younger than 3 years (n = 18) were usually located higher on principal component (PC) 1 than their diabetic counterparts (n = 17). PC1 accounts for 67% of the variability found in the data. The reverse was, however, true for controls older than 3 years (n = 9) with respect to their diabetic counterparts (n = 11). The average age values of the control–diabetic pairs were negatively correlated with their respective difference (control minus diabetic) on PC1 (control minus diabetic) (Fig. 1a) (p = 0.005, Spearman’s ρ test). When the difference between the control–diabetic pairs with regard to PC1 (67%) and PC4 (4%) was plotted (Fig. 1b), a complete separation of control–diabetic pairs at around the age of 3 years was observed. In Fig. 1b, pairs older than 2.9 years (age 2.93–4.6 years, mean 3.5 ± 0.45) clustered in the upper left quadrant, whereas pairs younger than 2.9 years (age 1.3–2.85 years, mean 2.1 ± 0.44) clustered in the lower right quadrant.

Difference between age-matched control–diabetic couples (controls minus diabetics) with respect to PC1 and PC4 at the 95% similarity level. (a) Difference between control–diabetic couples with respect to PC1 is negatively correlated with age. **p < 0.01 (Spearman’s ρ test). (b) Difference between control–diabetic couples with respect to PC1 and PC4 distinguishes couples younger than 2.9 years, represented by grey squares, from couples older than 2.9 years, represented by black circles. Dashed lines connecting the patient pairs are in sequential increasing order. The numbers 1.4, 2.8, 2.9 and 4.2 represent the ages of the youngest and oldest couple of each sequential order of the two age categories
This difference in the control and diabetic pairs can be mostly attributed to a shift in the abundance of species from Clostridium cluster XIVa as PC1 mostly describes the variation in abundance of Clostridium cluster XIVa (56%) (p = 2 × 10−10, Spearman’s ρ test). PC4 can in essence be best described by the abundance of Bacteroides + streptococci minus the combined abundance of Clostridium clusters I and XI (p = 6 × 10−15, Spearman’s ρ test) indicating a shift in prevalence between these groups. PC2 (8%) was most strongly correlated with the variation in abundance of Bifidobacteria (8.3%) (p = 2 × 10−11, Spearman’s ρ test), but neither was found to be correlated with the presence of diabetes or with age. PC3 (5%) was not correlated with either age or the presence of diabetes but mainly showed that Bacteroidetes (7.5%) and streptococci (7.3%) were negatively correlated with one another.
Redundancy and diversity analysis
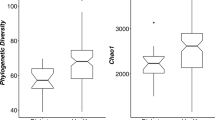
The separation of younger and older pairs was confirmed by redundancy analysis at the genus-like level as the older diabetic children formed a distinct subgroup as calculated using the Monte Carlo permutation test (p < 0.05; ESM Fig. 2). The microbiota of older diabetic children had an increased level of microbial richness compared with that of the other three groups (720 vs 611 ± 4) and also had a higher diversity (Fig. 2). Only a few phylogenetic groups differed in abundance between the faecal samples from the children with type 1 diabetes and the healthy controls if the samples were not separated into two age groups (Table 1).

Diversity analysis represented as a box plot. The whiskers represent the lowest and highest diversities measured within a particular group of samples. The boxes represent the interquartile ranges, and the lines within each box represents the median diversity value of a sample group. The Shannon index for diversity and the level of microbial richness are highest in diabetic children older than 2.9 years, especially when compared with non-diabetic children older than 2.9 years. *p < 0.05
Phylogenetic differences between the diabetic and control children with respect to age
In the age group of children younger than 2.9 years at the phylum level, Clostridium cluster XIVa was significantly more abundant in the control children compared with the age-matched diabetic children, whereas the samples from the diabetic children were more abundant in Bacteroidetes (Table 1). At the genus-like level, a higher prevalence of Lactobacillus plantarum et rel. was found in the controls, and a higher abundance of Streptococcus mitis et rel. in the children with diabetes (Table 1). Although the streptococci and Bacteroidetes were negatively correlated with one another (p = 0.02, Spearman’s ρ test, PC3), they were both positively correlated with PC4 (p = 0.01 and 0.001, respectively, Spearman’s ρ test). Most young diabetic children scored higher on PC4 than their control counterparts (Fig. 1b). Consequently, the sum of the abundances of Bacteroidetes and streptococci was positively associated with diabetes in children younger than 2.9 years (Table 1), indicating that the younger diabetic children often had a high abundance of either Bacteroidetes or streptococci (ESM Fig. 1). On the other hand, the sum of Clostridium clusters IV (11%) and XIVa, two dominant bacterial groups known to encompass most of the important butyrate-producing bacteria in the human gut ecosystem [27], was negatively associated with diabetes in pairs younger than 2.9 years in both a paired and an unpaired analysis (Table 1).
In the group of children older than 2.9 years, the largest difference between the control and the diabetic children was found within Clostridium cluster XIVa as a number of non-butyrate-producing species were more abundant in the older diabetic children (Table 1). Whereas the combined abundance of Clostridium clusters IV and XIVa in children younger than 2.9 years was higher in the controls than in the children with diabetes, this was not observed for children older than 2.9 years (Fig. 3a). Interestingly, the ratio between saccharolytic butyrate producers and saccharolytic bacteria that do not have butyrate as one of their main fermentation products within Clostridium clusters IV and XIVa (ESM Table 2) was significantly higher (p < 0.05) in the controls older than 2.9 years than in all the other groups (Fig. 3b). Apart from this, a slight overabundance of Clostridium stercorarium (0.6%) was found in older diabetic children (Table 1).

Microbial composition analysis of Clostridium clusters IV and XIVa. (a) The sum of the percentages of Clostridium clusters IV and XIVa in non-diabetic children younger than 2.9 years is higher than that in diabetic children younger than 2.9 years and non-diabetic children older than 2.9 years. *p < 0.05. (b) The fraction of butyrate-producing species within Clostridium clusters IV and XIVa is higher in non-diabetic children older than 2.9 years than in all other groups. † p < 0.05. The horizontal bars indicate the median values
Comparison between Finnish and non-Finnish diabetic children
In this study, a number of children were not Finnish (ESM Table 1). One of the control children was from Lithuania, but more importantly ten of the diabetic children were either from Estonia, France, Greece or Lithuania. To verify that the differences between the control and the diabetic children did not arise because a large proportion of the diabetic children (10/28) were not from Finland, a comparison was made between the Finnish and non-Finnish diabetic participants. The Finnish diabetic children had a significantly higher abundance of non-butyrate-producing species from Clostridium clusters IV and XIVa (Fig. 4; 41% vs 32%, p = 0.039). This could explain why the healthy controls with a young age (<2.9 years) had higher numbers of Clostridium cluster IV and XIVa (Fig. 3a), yet when analysing only the Finnish samples, the healthy Finnish controls had a greater number of Clostridium clusters IV and XIVa than the Finnish diabetic children (p = 0.008, Wilcoxon test). In addition, the sum of Bacteroides and streptococci, the second main pattern in children younger than 2.9 years, also remained significantly higher in the diabetic children in the above comparison (p = 0.021, Wilcoxon test). Furthermore, the presence of non-Finnish diabetic patients in the group of children older than 2.9 years cannot account for finding a higher ratio of butyrate to non-butyrate forming species in the healthy controls (Fig. 3b) as, in addition, non-Finnish diabetic individuals do not have a higher abundance of butyrate-producing species from Clostridium clusters IV and XIVa (19% vs 21%).

Finnish diabetic children have a higher abundance of non-butyrate-producing bacteria from Clostridium clusters IV and XIVa than non-Finnish diabetic children. Circles, children older than 2.9 years; squares, children younger than 2.9 years. *p < 0.05
In contrast, it was surprising to find that Bacteroides was no longer associated with type 1 diabetes in the children older than 2.9 years. This could indeed be explained by the introduction of non-Finnish diabetic patients. The reasoning for this is that the non-Finnish diabetic children had more clostridia of the closely related Clostridium clusters I and XI (7.5% vs 3.3%, p = 0.049), which are negatively correlated with the abundance of Bacteroides (p = 0.003). Five out of the ten pairs older than 2.9 years were non-Finnish, while this figure was only five out of 16 for the younger children. Hence, the use of non-Finnish diabetic children might have obfuscated the importance of Bacteroides with regard to type 1 diabetes in the pairs older than 2.9 years. Non-Finnish diabetic children furthermore had more enterococci (1.8% vs 0.05%, p = 0.0007) and more Eubacteria from Clostridium cluster XVI (0.28% vs 0.02%, p = 0.002), but these latter findings do not confound the conclusions. No difference in microbial diversity was observed between Finnish and non-Finish diabetic patients.
Discussion
The development of a healthy composition of intestinal microbiota occurs most rapidly in the first few years of life [3, 28]. This is most apparent in this study with respect to Clostridium clusters IV and XIVa in healthy controls (Fig. 3). It appears as if diabetic children are one ‘step’ behind in their gut microbial development (Fig. 3) and that this development goes in an aberrant direction (Fig. 2). A study by Yatsunenko et al showed that the phylogenetic bacterial composition evolves towards an adult-like configuration within a 3 year period after birth [29]. Our separation into two age categories of samples near the age of 3 (Fig. 1) aligns well with these results.
The rapid development of the gut microbiota during the first 3 years of life furthermore necessitates the use of paired analyses. In children older than 3 years, this requirement becomes less stringent and has the disadvantage of reducing the total number of measurements. Furthermore, two diabetic children older than 3 years were missing a paired control (as a matching sample failed to produce results). In order to use as many data points as possible, Table 1 shows the results from the paired and unpaired analyses in both age cohorts.
Clostridium clusters IV and XIVa were found to be similarly important in a recent study involving Finnish children that were positive for two or more diabetes-related autoantibodies, as several of their members were found to be negatively correlated with the number of diabetes-specific autoantibodies [11]. In a study of 7-year-old Spanish diabetic children who had already had type 1 diabetes for 4 years, the Blautia coccoides/Eubacterium rectale group (Clostridium cluster XIVa) was found to be less represented in diabetic children [12]. In the study by Brown et al, it was shown that the butyrate-producing genera Faecalibacterium (a member of Clostridium cluster IV) and Roseburia (a member of Clostridium cluster XIVa) were more predominant in healthy controls [15]. Several types of evidence exist on the importance of butyrate production with regard to the development of type 1 diabetes. Butyrate is not only the main energy source of the colonic epithelial cells [30], but also regulates the assembly of tight junctions and gut permeability [31], inhibits bacterial translocation [32] and has anti-inflammatory properties [18, 33]. Both gut permeability and inflammation have been linked to the development of type 1 diabetes in humans [14], and a reduced level of bacterial translocation is likely to protect against bacteria with diabetogenic properties, of which members of the Bacteroides fragilis group are prime suspects [8, 10–13].
The association of Bacteroidetes with diabetes has been reported both in humans and in animal models [8–13], but the association between streptococci and type 1 diabetes has only been reported in a study that was also carried out in very young children [15]. Even though the prevalence of streptococci was negatively correlated with that of Bacteroidetes, they were also found to be more prevalent in the younger diabetic children. Soft tissue infections with both streptococci and members of the Bacteroides fragilis group, as well as respiratory tract infections and bacteraemia associated with streptococci, are more common in diabetic patients [34, 35]. A possibly important similarity between Bacteroides and streptococci in their role in the development of type 1 diabetes is that they both produce glutamate decarboxylase, which might be the trigger for GAD autoimmunity via molecular mimicry [36]. In children who were positive for two or more diabetes-related autoantibodies the abundance of Bacteroides was most strongly linked with GAD autoimmunity (p = 0.01, M. de Goffau, unpublished observations) [11].
Contrary to expectations, bifidobacteria were not found to be correlated with diabetes in this study of new-onset diabetes, even though they have previously been found to be negatively associated with beta cell autoimmunity in Finnish children with two or more diabetes-related autoantibodies aged 4–14 years [11], in 7-year-old Spanish diabetic patients [12] and in a Turkish study with even older diabetic children [37]. Retaining a moderate bifidobacterial abundance as children grow older might be more important in relation to type 1 diabetes than the abundance of bifidobacteria in young children, whose bifidobacterial levels are likely to be still high enough overall. In several studies of type 1 diabetes, the Prevotella genus has also been found to be more prevalent in controls [10, 12, 13], although this was not found in the present study. The most logical reason for this is that Prevotella is simply rare in Finland even in healthy controls (0.6%). In the all-Finnish study by de Goffau et al, Prevotella was absent in 31 out of 36 children (M. de Goffau, unpublished observations) [11].
Because ten of the diabetic children were of non-Finnish origin whereas nearly all the control children were Finnish, it is critical to mention that the differences found between the diabetic and non-diabetic children with regard to the patterns related to Clostridium clusters IV and XIVa, as described in Fig. 3, have not been artificially introduced by possible differences in gut microbiota between Finland and the other European countries: analyses using only Finnish samples yield the same patterns. However, as non-Finnish diabetic children had a lower total abundance of non-butyrate-producing species from Clostridium clusters IV and XIVa, they had a higher abundance of several other groups, most importantly a higher abundance of the closely related Clostridium clusters I and XI [38, 39], which are found to be strongly negatively correlated with Bacteroides. As a relatively large proportion of the diabetic children older than 2.9 years did not come from Finland, this might have obfuscated the correlation between Bacteroides and type 1 diabetes in this age category.
As samples were sent at ambient temperatures before being stored at −75°C, it is not inconceivable that this might have had a small effect on the faecal microbial composition during transit [40, 41]. However, as the above is true for all the case and control samples, it is impossible that this might have influenced the comparison between cases and controls with respect to the observed differences.
The causality and even the direction of causality of the changes in the microbial composition reported in this and other similar studies still need to be substantiated by functional studies. Predictions about, for example, the potential for butyrate production based on 16S rRNA sequence frequencies will likely remain at best tentative as the actual butyrate production of the species known to be capable of doing this is highly dependent on external factors such as the diet and the presence and abundance of other bacterial species. Confirming predictions about short-chain fatty acid production profiles is furthermore likely to remain difficult as the majority of short-chain fatty acids are taken up by the colon [42] and because of the volatility of such compounds during transit and handling. In vitro co-culture experiments will, however, help to validate the importance and existence of bacterial cross-feeding complexes.
In conclusion, although distinct differences have been found in each age category between the healthy and diabetic children, the main differences with regard to Clostridium clusters IV and XIVa appear to represent two sides of the same coin, as they together emphasise the importance of developing balanced bacterial cross-feeding complexes that have sufficient potential for butyrate formation. Dietary interventions aimed at achieving or maintaining optimal butyrate production levels might measurably reduce the risk of developing type 1 diabetes, especially in children with high-risk HLA genotypes.
Abbreviations
- HITChip:
-
Human Intestinal Tract Chip
- PC:
-
Principal component
- PCA:
-
Principal component analysis
References
Borody TJ, Khoruts A (2012) Fecal microbiota transplantation and emerging applications. Nat Rev Gastroenterol Hepatol 9:88–96
de Vos WM, de Vos EA (2012) Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr Rev 70(Suppl 1):S45–S56
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489:220–230
Neish AS (2009) Microbes in gastrointestinal health and disease. Gastroenterology 136:65–80
Surana NK, Kasper DL (2012) The yin yang of bacterial polysaccharides: lessons learned from B. fragilis PSA. Immunol Rev 245:13–26
Kosiewicz MM, Zirnheld AL, Alard P (2011) Gut microbiota, immunity, and disease: a complex relationship. Front Microbiol 2:180
Sokol H, Pigneur B, Watterlot L et al (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 105:16731–16736
Brugman S, Klatter FA, Visser JT et al (2006) Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 49:2105–2108
Wen L, Ley RE, Volchkov PY et al (2008) Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455:1109–1113
Giongo A, Gano KA, Crabb DB et al (2011) Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 5:82–91
de Goffau MC, Luopajarvi K, Knip M et al (2013) Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes 62:1238–1244
Murri M, Leiva I, Gomez-Zumaquero JM et al (2013) Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med 11:46
Mejía-León ME, Petrosino JF, Ajami NJ, Domínguez-Bello MG, de la Barca AM (2014) Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci Rep 4:3814
Vaarala O, Atkinson MA, Neu J (2008) The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes 57:2555–2562
Brown CT, Richardson AG, Giongo A et al (2011) Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One 6:e25792
Van Immerseel F, Ducatelle R, de Vos M et al (2010) Butyric acid-producing anaerobic bacteria as a novel probiotic treatment approach for inflammatory bowel disease. J Med Microbiol 59:141–143
Flint HJ, Duncan H, Scott KP, Louis P (2007) Interactions and competition within the microbial community of the human colon: links between diet and health. Environ Microbiol 9:1101–1111
Segain JP, de la Raingeard BD, Bourreille A et al (2000) Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 47:397–403
Furet JP, Kong LC, Tap J et al (2010) Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes 59:3049–3057
Onkamo P, Vaananen S, Karvonen M, Tuomilehto J (1999) Worldwide increase in incidence of type I diabetes—the analysis of the data on published incidence trends. Diabetologia 42:1395–1403
Karvonen M, Pitkaniemi J, Tuomilehto J (1999) The onset age of type 1 diabetes in Finnish children has become younger. The Finnish Childhood Diabetes Registry Group. Diabetes Care 22:1066–1070
Weets I, de Leeuw I, Du Caju MV et al (2002) The incidence of type 1 diabetes in the age group 0–39 years has not increased in Antwerp (Belgium) between 1989 and 2000: evidence for earlier disease manifestation. Diabetes Care 25:840–846
Pundziute-Lycka A, Dahlquist G, Nystrom L et al (2002) The incidence of type I diabetes has not increased but shifted to a younger age at diagnosis in the 0–34 years group in Sweden 1983–1998. Diabetologia 45:783–791
Rajilic-Stojanovic M, Heilig HG, Molenaar D et al (2009) Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol 11:1736–1751
Yu Z, Morrison M (2004) Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36:808–812
Jalanka-Tuovinen J, Salonen A, Nikkila J et al (2011) Intestinal microbiota in healthy adults: temporal analysis reveals individual and common core and relation to intestinal symptoms. PLoS One 6:e23035
Lous P, Flint HJ (2009) Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett 294:1–8
Koenig JE, Spor A, Scalfone N et al (2011) Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 108(Suppl 1):4578–4585
Yatsunenko T, Rey FE, Manary MJ et al (2012) Human gut microbiome viewed across age and geography. Nature 486:222–227
Hague A, Butt AJ, Paraskeva C (1996) The role of butyrate in human colonic epithelial cells: an energy source or inducer of differentiation and apoptosis? Proc Nutr Soc 55:937–943
Peng L, Li ZR, Green RS, Holzman IR, Lin J (2009) Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr 139:1619–1625
Lewis K, Lutgendorff F, Phan V, Soderholm JD, Sherman PM, McKay DM (2010) Enhanced translocation of bacteria across metabolically stressed epithelia is reduced by butyrate. Inflamm Bowel Dis 16:1138–1148
Russo I, Luciani A, de Cicco P, Troncone E, Ciacci C (2012) Butyrate attenuates lipopolysaccharide-induced inflammation in intestinal cells and Crohn’s mucosa through modulation of antioxidant defense machinery. PLoS One 7:e32841
Joshi N, Caputo GM, Weitekamp MR, Karchmer AW (1999) Infections in patients with diabetes mellitus. N Engl J Med 341:1906–1912
Nowakowska M, Jarosz-Chobot P (2004) Streptococcus group B (GBS)—characteristic, occurrence in children and adolescents with type 1 diabetes mellitus. Pol J Microbiol 53:17–22
García E, López R (1995) Streptococcus pneumoniae type 3 encodes a protein highly similar to the human glutamate decarboxylase (GAD65). FEMS Microbiol Lett 133:113–119
Soyucen E, Gulcan A, Aktuglu-Zeybek AC, Onal H, Kiykim E, Aydin A (2013) Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatr Int. doi:10.1111/ped.12243
Collins MD, Lawson PA, Willems A et al (1994) The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol 44:812–826
Johnson JL, Francis BS (1975) Taxonomy of the Clostridia: ribosomal ribonucleic acid homologies among the species. J Gen Microbiol 88:229–244
Cardona S, Eck A, Cassellas M et al (2012) Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol 12:158
Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N (2010) Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett 307:80–86
Nyman M (2000) Fermentation and bulking capacity of indigestible carbohydrates: the case of inulin and oligofructose. Br J Nutr 87(Suppl 2):S163–S168
Acknowledgements
D. Dimitripoulou (University Medical Center Groningen, Groningen, the Netherlands) and H. Heilig (Wageningen University, Wageningen, the Netherlands) are acknowledged for their technical assistance. The collection of study series was carried out in collaboration with several investigators as described below: The Finnish Type 1 Diabetes Prediction and Prevention study (DIPP) investigators: R. Veijola (University of Oulu, Oulu, Finland), O. Simell, T. Simell, J. Toppari, J. Ilonen (University of Turku, Turku, Finland), M. Knip (University of Helsinki, Helsinki, Finland) and H. Hyöty (University of Tampere, Tampere, Finland); the Viruses in Diabetes (VirDiab) study investigators: H. Hyöty, S. Oikarinen, K. Kankaanpää (University of Tampere), D. Hober, B. Lucas, J. Weill, C. Stuckens, G.-A. Loeuille (University Lille 2 and CHRU Lille, Lille, France and CH Dunkerque, Dunkerque, France), P. Muir (Kings College London, London, UK), K. Taylor (Queen Mary & Westfield College, London, UK), G. Stanway, C. Williams (University of Essex, Colchester, UK), C. S. Bartsocas, A. Vazeou, E. Bozas, M. Dokopoulou, D. Delis (National University of Athens, Greece), J. Ludvigsson (Linköping University, Sweden), P. Keskinen, M.-T. Saha (Tampere University Hospital), J. Ilonen (University of Turku), M. Knip (University of Helsinki), D. Marciulionyte (Kaunas University of Medicine, Kaunas, Lithuania), V. Tillman, K. Heilman and R. Uibo (University of Tartu, Tartu, Estonia).
Funding
This study was supported by the Competitive Research Funding of the Tampere University Hospital, the JDRF, the Academy of Finland and the European Commission (VIRDIAB Project, Quality of Life Programme, KA2, contract number QLK 2-CT-2001-01910 and the European Union Seventh Framework Package FP7/2007-2013 (DIABIMMUNE project).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
MCG, BB and HaH contributed to the acquisition of research data. MCG and SF performed the statistical analyses. HaH and HeH provided the study materials. HaH wrote the sample collection section, and BB and SF wrote the HITChip analysis section. MCG wrote the rest of the manuscript. WMV contributed to developing, improving and organising the HITChip and its pipeline. All authors contributed to the discussion. HeH and GWW initiated the study and were responsible, together with HJMH and WMV, for the design of the study. BB, GWW, WMV, HeH and HJMH reviewed and edited the manuscript. All authors gave final approval to the manuscript version to be published. HJMH is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 16 kb)
ESM Table 2
(PDF 27 kb)
ESM Fig. 1
(PDF 898 kb)
ESM Fig. 2
(PDF 51.8 kb)
Rights and permissions
About this article

Cite this article
de Goffau, M.C., Fuentes, S., van den Bogert, B. et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia 57, 1569–1577 (2014). https://doi.org/10.1007/s00125-014-3274-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3274-0