
Abstract
Aim/hypothesis
Hyperglucagonaemia is a characteristic of several clinical conditions (e.g. end-stage renal disease (ESRD), type 2 diabetes, obesity before and after Roux-en-Y gastric bypass (RYGB) and vagotomy with pyloroplasty), but the molecular nature of ‘immunoreactive’ glucagon is poorly characterised. The specific determination of fully processed, intact glucagon requires a ‘sandwich’ assay employing a combination of antibodies directed against both N- and C-termini. We compared a novel assay for intact glucagon with a highly sensitive C-terminal RIA (hitherto considered specific) to determine the extent to which the hyperglucagonaemia measured in clinical samples was caused by authentic glucagon.
Methods
We examined the performance of three commercial glucagon ‘sandwich’ ELISAs. The ELISA with the best overall performance was selected to compare glucagon measurements in clinical samples with an established glucagon RIA.
Results
The first assay performed poorly: there was high cross-reactivity with glicentin (22%) and a lack of sensitivity for glucagon. The second and third assays showed minor cross-reactivity (1–5%) with oxyntomodulin and glicentin; however, the second assay had insufficient sensitivity for glucagon in plasma (>10–20 pmol/l). Thus, only the third assay was suitable for measuring glucagon concentrations in clinical samples. The ELISA and RIA measured similar glucagon levels in healthy individuals. Measurements of samples from individuals with abnormally high (type 2 diabetes or obese) or very elevated (post vagotomy with pyloroplasty, post-RYGB) glucagon levels were also similar in both assays. However, glucagon levels in participants with ESRD were much lower when measured by ELISA than by RIA, indicating that the apparent hyperglucagonaemia is not caused by fully processed intact glucagon.
Conclusions/interpretation
For most purposes, sensitive C-terminal glucagon RIAs are accurate. However, measurements may be spuriously high, at least in patients with renal disease.
Trial Registration Samples from type 2 diabetic and normoglucose-tolerant patients before and 1 year after RYGB were from a study by Bojsen-Møller et al (trial registration number NCT 01202526). Samples from vagotomised and control individuals were from a study by Plamboeck et al (NCT01176890). Samples from ESRD patients were from a study by Idorn et al (NCT01327378).
Similar content being viewed by others


Introduction
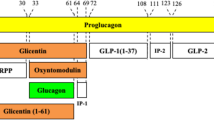
Glucagon, a cleavage product of proglucagon, has an important role in the pathology of diabetes [1, 2]: increased secretion (hyperglucagonaemia) and impaired glucose-mediated suppression of glucagon are both thought to contribute to diabetic hyperglycaemia [3–6]. Elevated plasma glucagon levels have also been reported after Roux-en-Y gastric bypass (RYGB) or vagotomy (plus pyloroplasty), or in patients with end-stage renal disease (ESRD) [7–9]. Paradoxically, glucagon levels are generally elevated at elevated glucose levels that should suppress glucagon secretion. The cause and molecular nature of this elevated ‘immunoreactive’ glucagon have, therefore, been questioned.
There are inherent problems in glucagon measurement regarding sensitivity and specificity. Antibodies specific to the free C-terminus of glucagon will react with glucagon [10], but not with glicentin or oxyntomodulin, and are reported to specifically detect fully processed pancreatic glucagon. While this may be a valid assumption when measuring glucagon in healthy individuals, such antibodies will also detect N-terminally extended or truncated forms of glucagon (e.g. glucagon 18–29, 19–29, 3–29 and 1–61 [11, 12]), the concentrations of which may be altered in pathological conditions that affect either the secretion or clearance of proglucagon-derived peptides. The specific determination of fully processed, intact glucagon might therefore be accomplished using a ‘sandwich’ assay employing a combination of N- and C-terminal anti-glucagon antibodies. The purpose of the present study was to evaluate three recently available sandwich ELISAs, and then to compare the performance of this type of assay with an established RIA in measuring glucagon levels in plasma samples from patients in whom hyperglucagonaemic had previously been identified.
Methods
Commercial glucagon assays
Three commercial sandwich ELISA kits from Mercodia, Meso Scale Discovery and Millipore were tested (Table 1). Kits were stored and used according to the manufacturers’ recommendations, and three different lots were tested for each kit. Mercodia kits were a gift from the manufacturer (Mercodia A/S, Uppsala, Sweden).
Synthetic peptides
The indicated amounts of synthetic glucagon 1–29 (H-6790; Bachem, Bubendorf, Switzerland), oxyntomodulin (H-6058; Bachem) and glicentin (custom made; service no. SC1208; GenScript, Piscataway, USA) were verified as described previously [13], by quantitative amino acid analysis (QAAA) and an in-house mid-region specific glucagon assay (antiserum no. 4304; University of Copenhagen, Copenhagen, Denmark) [10].
Assay performance
Assays were characterised as previously described [13]. Specificity was determined using recovery experiments. Briefly, known amounts of each peptide (0–150 pmol/l) were added to the assay buffer supplied with each kit (=spiked buffer) or to plasma (pooled from nine healthy volunteers; collected into EDTA containing 500 kallikrein inhibitor effect (KIE)/ml aprotinin; [Bayer Health Care AG, Leverkusen, Germany]; = spiked plasma). For each assay, spiked buffer or spiked plasma samples were measured in duplicate on three separate occasions using three separate kits.
Sensitivity was estimated by determining the lowest peptide concentrations determined to be significantly different from zero (by paired analysis of three duplicate assays). Precision was determined by spiking a single pooled plasma preparation with 0, 1, 2, 5, 10 or 20 pmol/l glucagon, and assaying each concentration in replicates of five to six in a single experiment. Means and SDs were calculated for each glucagon concentration.
Measurement of clinical samples
An in-house RIA employing C-terminal glucagon-specific antiserum (no. 4305; University of Copenhagen, Copenhagen, Denmark), previously characterised for sensitivity, specificity and precision, was used to measure glucagon [10, 13–15]. Clinical plasma samples were extracted with ethanol to remove nonspecific cross-reacting factors, resulting in 74% ± 4% (mean ± SD) glucagon recovery. The sensitivity of this RIA is approximately 1 pmol/l and the intra-assay CV is <6%. Identical plasma samples were also assayed by sandwich ELISA according to the manufacturers’ instructions, including the use of unextracted plasma samples where recommended. This assay was performed using the commercial ELISA determined to have the best specificity and sensitivity.
Patient samples
Stored plasma samples from three previously published clinical studies were examined. Clinical studies were conducted in accordance with the latest revision of the Helsinki Declaration, approved by the Scientific-Ethical Committee of the Capital Region of Denmark and by the Danish Data Protection Agency, and registered at ClinicalTrials.gov.
Plasma samples from type 2 diabetes patients and individuals with normal glucose tolerance (NGT) before and 1 year after RYGB were obtained from Bojsen-Møller et al (trial registration number NCT 01202526) [7]; plasma samples from vagotomised and control individuals were obtained from Plamboeck et al (NCT01176890) [8]; and plasma samples from ESRD patients were obtained from Idorn et al (NCT01327378) [9].
Statistical analysis
To evaluate assay specificity, concentrations of glucagon, oxyntomodulin or glicentin measured in each assay were plotted against the calculated concentrations (after subtraction of plasma only values), and linear regression analyses were performed. The regression coefficient r 2 shows the fit of data to the line, and the slopes of the best fit lines correspond to recovery in the assay (100% recovery equates to full cross-reactivity, while a large regression coefficient indicates that findings were consistent and reproducible over the full test range). For the slopes of each line for each peptide and each assay, p values were calculated for the null hypothesis (i.e. the horizontal line). To evaluate precision, a one-way ANOVA for repeated measurements followed by Bonferroni post hoc analysis was performed to compare the spiked samples with the negative control (unspiked human plasma).
For clinical samples, two-way ANOVA was used to determine significant differences between assays during either ingestion of a meal or an OGTT challenge in the various patient groups. The AUC was calculated using the trapezoidal rule. Total C-terminal glucagon immunoreactivity (by in-house RIA) was compared with intact glucagon measurements (by ELISA) using a paired t test. p < 0.05 was considered statistically significant. All calculations were performed using GraphPad Prism (version 5.0; La Jolla, CA, USA).
Results
Synthetic peptide verification
Peptide concentration, expressed as a percentage of the expected concentration, was determined for each synthetic peptide: glucagon 1–29 (RIA 95%, QAAA 92%), oxyntomodulin (RIA 67%, QAAA 74%) and glicentin (RIA 75%, QAAA 78%).
Evaluation of commercial ELISA kits
The specificity of three commercial kits was evaluated by the recovery of spiked glucagon, oxyntomodulin or glicentin in buffer (Table 2) and human plasma (Table 3). The Meso Scale Discovery assay had relatively poor specificity: recoveries of 7–12% were found for both oxyntomodulin and glicentin in buffer; and in human plasma, oxyntomodulin recovery was lower (around 5%), but glicentin recovery was higher (~22%). Furthermore, glucagon recoveries were variable between assay runs in each matrix, and were also influenced by the matrix. Thus, the Meso Scale Discovery assay tended to overestimate the glucagon concentration in buffer, while the concentration in plasma was underestimated by around one-third. Although the specificities of the Millipore and Mercodia sandwich ELISAs were better, both showed some cross-reactivity with oxyntomodulin (<5%) and glicentin (<2%) in both buffer and plasma. For the Millipore kit, full recovery of glucagon in buffer was obtained, but plasma had a modest negative effect, with recoveries falling to ~75%. However, for this assay, results were consistent between runs. Results obtained with the Mercodia kit were also reproducible between assay runs using kits with differing lot numbers, and full recovery of glucagon was obtained from both buffer and plasma.
Assay sensitivity and precision varied between the three kits (Table 4). Background immunoreactivity in the negative control differed among kits: similar values were measured with the Millipore and Mercodia kits, whereas approximately twofold higher levels were obtained with the Meso Scale Discovery kit. The Meso Scale Discovery kit had poor precision and sensitivity, and it failed to discriminate between samples containing different amounts of glucagon. Although the average measured concentrations tended to increase (from 11 pmol/l in the negative control to 19 pmol/l for the 20 pmol/l glucagon sample, i.e. a nominal increase of 8 pmol/l), the SDs also increased (from 5 to 9 pmol/l), indicating a lack of precision and, therefore, sensitivity in the physiological range of glucagon (1–20 pmol/l). Both Millipore and Mercodia kits had better precision. For Millipore, samples containing up to 5 pmol/l glucagon could not be discriminated from the negative control, and the difference in measurements for samples containing between 0 and 10 pmol/l was registered as 6 pmol/l. However, correct measurements were obtained for samples containing 20 pmol/l glucagon. Although the Mercodia kit appeared to detect 1 pmol/l glucagon, the same values were obtained for samples containing 1 and 2 pmol/l glucagon. However, concentrations of 5 pmol/l and above were correctly registered.
This analysis indicated that the Mercodia assay has the best performance, in terms of specificity, precision and sensitivity data. This assay was therefore chosen for comparison with the standard RIA.
Clinical samples
In healthy participants (Fig. 1a, b), similar results for glucagon levels were obtained with the in-house C-terminal RIA (detecting C-terminal glucagon) and the sandwich ELISA (detecting intact glucagon). Equivalent overall glucagon excursions (by AUC) were measured in response to a standardised meal challenge. In vagotomised patients (Fig. 1c), there was a trend toward lower basal measurements by sandwich ELISA (5 vs 9 pmol/l; p = 0.066), and for slightly lower measurements for intact glucagon than for C-terminal glucagon during the late phase of the meal challenge (at 120 min, p = 0.07; at 180 min, p < 0.05). However, the overall AUC was not significantly different between assays (Fig. 1d), even though higher glucagon levels were measured than for matched control participants (Fig. 1b). In contrast, in patients with ESRD, C-terminal glucagon measurements were significantly higher than intact glucagon measurements (Fig. 1e–h), both in the basal state and in response to both OGTT (Fig. 1e, f) and isoglycaemic i.v. glucose infusion (IIGI; Fig. 1g, h).

Total glucagon measured by a C-terminal RIA (circle, solid line) and intact glucagon measured by a sandwich ELISA (square, dashed line) plotted against time (a, c, e, g) or represented by computed AUC (b, d, f, h). Healthy participants (a, b) and vagotomised patients (c, d) were stimulated with a standardised meal test. ESRD patients were either stimulated with OGTT (e, f) or IIGI (g, h). *p < 0.01 indicates significant changes in AUC by a paired student t test or two-way ANOVA. n = 8 for all groups; data are presented as mean ± SD
Patients with type 2 diabetes have also been reported to exhibit hyperglucagonaemia. In the present study, there was a trend (p = 0.08) for oral glucose-stimulated glucagon levels measured by in-house RIA to be higher in type 2 diabetes patients (Fig. 2a, b) than in obese participants with NGT (Fig. 2c, d). However, within each group, measurements of intact and C-terminal glucagon were similar.

Total glucagon measured by a C-terminal RIA (circle, solid line) and intact glucagon measured by sandwich ELISA (square, dashed line) plotted against time (a, c, e, g) or represented by computed AUC (b, d, f, h). Type 2 diabetes patients before (a, b) and after RYGB surgery (e, f) during an OGTT challenge. NGT patients before (c, d) and after (g, h) RYGB surgery during an OGTT challenge. n = 8 for all groups; data are presented as mean ± SD
In the same individuals studied 1 year after RYGB, there was a trend (at 30 min, p = 0.09; at 60 min, p = 0.08) for type 2 diabetes patients to have lower levels of intact glucagon than C-terminal glucagon, but the overall glucagon response to an OGTT was similar for both assays (Fig. 2e, f). In obese NGT individuals, the glucagon response to an OGTT was slightly higher (p = 0.11) after RYGB, but there was no difference in measurements between the two assays (Fig. 2g, h).
Discussion
Hyperglucagonaemia is a characteristic feature of type 2 diabetes that has attracted considerable interest because suppression of glucagon secretion is a potential therapeutic aim [2, 16]. However, hyperglucagonaemia is also observed in renal disease and obesity, and after operations that alter the gastrointestinal delivery of nutrients, such as RYGB and vagotomy plus pyloroplasty [7–9]. The immunoreactive glucagon seen in these hyperglucagonaemic states has hitherto been poorly characterised, but the elevated levels may result from increased levels of authentic pancreatic glucagon (arising from increased secretion and/or reduced clearance) and/or the accumulation of other moieties containing the antigenic determinant of glucagon (e.g. other intestinal products of the GCG gene [encoding proglucagon], such as glicentin or oxyntomodulin, or metabolites arising from glucagon degradation). For instance, Baldissera and Holst reported increased concentrations of N-terminally elongated glucagon-containing peptides (including proglucagon 1–61) in uraemic pigs and pancreatectomised humans in the 1990s [11, 17, 18]. In addition, hyperglucagonaemia seen in type 2 diabetes is suggested to be secondary to the loss of endogenous insulin-induced suppression of alpha cell secretion [19]. Until recently, most pancreatic glucagon assays (including our in-house RIA) have relied on C-terminal antibodies; however, such assays can detect any molecule containing this structural element, including N-terminally elongated or truncated forms of glucagon. In contrast, a sandwich ELISA using a combination of antibodies that specifically recognise unmodified N- and C-terminal epitopes should not detect these molecules.
In the present study, we investigated the accuracy of hyperglucagonaemia findings in various disease states using one of the new sandwich ELISAs [20] that detect only intact glucagon in order to distinguish increased pancreatic glucagon from apparent hyperglucagonaemia resulting from cross-reactive peptides. We first tested three commercially available glucagon sandwich ELISAs for specificity toward full-length glucagon (Tables 2 and 3), and evaluated whether they exhibited the required sensitivity for measuring biological samples in the low pmol/l physiological range (Table 4). The Meso Scale Discovery kit showed considerable cross-reactivity with other proglucagon-derived peptides, whereas the Millipore and Mercodia kits were more specific, detecting mainly glucagon (Tables 2 and 3). They also showed greater sensitivity (Table 4), although for the Millipore assay, precision at lower concentrations was not ideal. Glucagon concentrations typically reach levels near or below 1 pmol/l during glucose or insulin suppression, and the Millipore assay has inadequate sensitivity in this range. However, further evaluation of this kit would require clinical samples taken after suppression of glucagon levels. Therefore, the Mercodia assay [20] was selected for the present study. Its lack of reactivity with elongated glucagon forms suggests a high degree of specificity, although it may still react with truncated glucagon peptides. These could arise, for example, after cleavage by dipeptidyl peptidase-4 (DPP-4), which would theoretically generate glucagon 3–29; however, glucagon does not appear to be a good substrate for DPP-4 in vivo [21]. Alternatively, truncated peptides could result from glucagon degradation by neutral endopeptidase 24.11 [12]. Such metabolites may be detected by the C-terminal RIA; however, Mercodia have stated that their assay does not cross-react with glucagon 3–29. Accuracy can also be affected by the matrix, particularly for unextracted plasma samples. Nonspecific detection of plasma components may lead to high levels of inter-subject variability. A limitation of the present study is, therefore, the relatively small numbers of individuals examined, although the error bars (SD) were generally small.
In healthy individuals, glucagon concentrations were low, and similar values were obtained for both the RIA (employing ethanol-extracted plasma to minimise nonspecific interference) and the ELISA, suggesting that the immunoreactive glucagon was composed entirely of authentic glucagon. In comparison, participants with vagotomy plus pyloroplasty had paradoxical hyperglucagonaemia during the meal challenge, although concentrations measured in both assays were again similar. Vagotomy plus pyloroplasty is associated with greatly accelerated gastric emptying and increased secretion of proglucagon products, including glucagon-like peptide 1 (GLP-1), from intestinal L-cells [22]. However, given that the assays measure authentic glucagon, rather than other proglucagon-derived peptides, it is likely that an intestinal factor stimulates the release of glucagon from pancreatic alpha cells. This factor may be glucose-dependent insulinotropic polypeptide (GIP), which is glucagonotropic [23] and shows increased release after vagotomy plus pyloroplasty [8]. However, the source could also be the gut itself. Elevated glucagon immunoreactivity has been observed in humans following total pancreatectomy, and chromatographic analysis has shown that the immunoreactive component has a similar size to glucagon [11, 17, 18]. In other words, glucagon might be formed by pancreatic-type processing of proglucagon in intestinal L-cells after pancreatectomy, and a similar mechanism could operate after vagotomy plus pyloroplasty. Patients with RYGB show a similar increase in the release of proglucagon products from the gut (but do not generally hypersecrete GIP); however, once again the ELISA and C-terminal RIA gave similar results, suggesting that postprandial hyperglucagonaemia in these individuals may therefore be a consequence of altered proglucagon processing in the gut, leading to the formation of fully processed glucagon. In contrast, in patients with ESRD, glucagon concentrations measured by RIA were significantly higher than those determined by ELISA, which remained within the normal range. Thus, hyperglucagonaemia in these patients is not caused by authentic glucagon. It has been suggested that circulating concentrations of N-terminally elongated forms of glucagon, perhaps including proglucagon 1–61 [11, 17, 18], are elevated in renal disease. These moieties are biologically inactive (at least with respect to effects on hepatic glucose output), which may also explain why the marked ‘hyperglucagonaemia’ does not translate into a diabetic metabolism in these patients [9]. As expected, basal and postprandial glucagon levels in type 2 diabetes patients were also increased in the present study. This was shown in both assays, thereby confirming that diabetic hyperglucagonaemia is caused by authentic glucagon and supporting conclusions drawn from mimicry studies in healthy humans [24] that inappropriately raised glucagon levels contribute to metabolic disturbances in type 2 diabetes.
In conclusion, the present study provides evidence that the hyperglucagonaemia previously observed in type 2 diabetes patients and after surgical operations associated with accelerated gastric emptying (including vagotomy with pyloroplasty and RYGB) is caused by elevated levels of intact, fully processed glucagon. Our studies do not, however, indicate whether this glucagon originates from the gut or the pancreas, although this information is important for our understanding of the pathophysiology of these conditions. In contrast, although patients with renal disease have apparent hyperglucagonaemia, this is not caused by elevated levels of authentic glucagon and further studies are required to identify the immunoreactive factor. Therefore, sensitive C-terminal glucagon RIAs will be reasonably accurate for most purposes, but may give spuriously high measurements when renal function is impaired.
Abbreviations
- ESRD:
-
End-stage-renal disease
- GLP:
-
Glucagon-like peptide
- NGT:
-
Normal glucose tolerance
- QAAA:
-
Quantitative amino acid analysis
- RYGB:
-
Roux-en-Y gastric bypass
References
Bell GI, Santerre RF, Mullenbach GT (1983) Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature 302:716–718
Holst JJ, Christensen M, Lund A et al (2011) Regulation of glucagon secretion by incretins. Diabetes Obes Metab 13(Suppl 1):89–94
Ohneda A, Ohneda M (1988) Effect of glicentin-related peptides upon the secretion of insulin and glucagon in the canine pancreas. Tohoku J Exp Med 155:197–204
Brand CL, Jorgensen PN, Svendsen I, Holst JJ (1996) Evidence for a major role for glucagon in regulation of plasma glucose in conscious, nondiabetic, and alloxan-induced diabetic rabbits. Diabetes 45:1076–1083
Unger RH, Aguilar-Parada E, Muller WA, Eisentraut AM (1970) Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 49:837–848
Toft-Nielsen MB, Madsbad S, Holst JJ (2001) Determinants of the effectiveness of glucagon-like peptide-1 in type 2 diabetes. J Clin Endocrinol Metab 86:3853–3860
Bojsen-Møller KN, Dirksen C, Jørgensen NB et al (2014) Early enhancements of hepatic and later of peripheral insulin sensitivity combined with increased postprandial insulin secretion contribute to improved glycemic control after Roux-en-Y gastric bypass. Diabetes 63:1725–1737
Plamboeck A, Veedfald S, Deacon CF et al (2013) The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am J Physiol Gastrointest Liver Physiol 304:G1117–G1127
Idorn T, Knop FK, Jorgensen M, Holst JJ, Hornum M, Feldt-Rasmussen B (2013) Gastrointestinal factors contribute to glucometabolic disturbances in nondiabetic patients with end-stage renal disease. Kidney Int 83:915–923
Holst JJ (1982) Evidence that enteroglucagon (II) is identical with the C-terminal sequence (residues 33–69) of glicentin. Biochem J 207:381–388
Baldissera FG, Holst JJ (1986) Glicentin 1–61 probably represents a major fraction of glucagon-related peptides in plasma of anaesthetized uraemic pigs. Diabetologia 29:462–467
Trebbien R, Klarskov L, Olesen M, Holst JJ, Carr RD, Deacon CF (2004) Neutral endopeptidase 24.11 is important for the degradation of both endogenous and exogenous glucagon in anesthetized pigs. Am J Physiol Endocrinol Metab 287:E431–E438
Bak MJ, Albrechtsen NW, Pedersen J et al (2014) Specificity and sensitivity of commercially available assays for glucagon and oxyntomodulin measurement in humans. Eur J Endocrinol 170:529–538
Holst JJ (1980) Evidence that glicentin contains the entire sequence of glucagon. Biochem J 187:337–343
Bak MJ, Albrechtsen NW, Hartmann B et al (2014) Effect of aprotinin (Trasylol™) on degradation of exogenous and endogenous glucagon in human, mouse and rat plasma. J Endocrinol Diabetes 1:5
Holst JJ, Knop FK, Vilsboll T, Krarup T, Madsbad S (2011) Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care 34:S251–S257
Holst JJ, Pedersen JH, Baldissera F, Stadil F (1983) Circulating glucagon after total pancreatectomy in man. Diabetologia 25:396–399
Holst JJ (1983) Gut glucagon, enteroglucagon, gut glucagonlike immunoreactivity, glicentin—current status. Gastroenterology 84:1602–1613
Meier JJ, Ueberberg S, Korbas S, Schneider S (2011) Diminished glucagon suppression after β-cell reduction is due to impaired α-cell function rather than an expansion of α-cell mass. Am J Physiol Endocrinol Metab 300:E717–E723
Gäreskog MCA, Schwanbeck M, Karamihos E et al (2013) Development and validation of a high-sensitivity ELISA forspecific measurement of glucagon in rat and mouse samples. IDF 2013 Abstract PD-0657
Deacon CF, Kelstrup M, Trebbien R, Klarskov L, Olesen M, Holst JJ (2003) Differential regional metabolism of glucagon in anesthetized pigs. Am J Physiol Endocrinol Metab 285:E552–E560
Plamboeck A, Veedfald S, Deacon CF et al (2013) Characterisation of oral and i.v. glucose handling in truncally vagotomised subjects with pyloroplasty. Eur J Endocrinol 169:187–201
Meier JJ, Gallwitz B, Siepmann N et al (2003) Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 46:798–801
Shah M, Law JH, Micheletto F et al (2014) Contribution of endogenous glucagon-like peptide 1 to glucose metabolism after Roux-en-Y gastric bypass. Diabetes 63:483–493
Acknowledgements
The authors would like to thank Mercodia A/S (Sylveniusgatan 8A, SE-754 50, Uppsala, Sweden) for providing assays for clinical glucagon measurements. We also wish to thank L. Albæk and S. Pilgaard of the Department for Translational Metabolism, University of Copenhagen, Denmark, for outstanding technical assistance. A special thanks also goes to R. Kuhre of the Department for Translational Metabolism, University of Copenhagen, Denmark, for many fruitful discussions.
Funding
Academic staff and students at the University of Copenhagen were supported by the NNF Centre for Basic Metabolic Research, University of Copenhagen, The Danish Council for Independent Research (DFF–1333–00206A) and the Faculty of Health Sciences Foundation.
Duality of interest
JJH has provided two online lectures for Mercodia and discussed the principles behind peptide hormone measurements by sandwich ELISAs (compensation was received for one of these, a ‘webinar’), but has not otherwise received any honoraria, grants, lecture/other fees, shareholding or similar financial benefits from Mercodia. Otherwise, the authors declare that there is no duality of interest associated with this manuscript. Mercodia provided assay kits without imposing restrictions on the study design or interpretation of results.
Contribution statement
NWA, BH, SV, JAW, AP, KBM, TI, BFR, FKK, TV, SM, CFD and JJH provided substantial contribution to the concept and design. NWA, BH, JAW, CFD and JJH substantially contributed to data analysis and interpretation. NWA, CFD and JJH drafted the manuscript. BH, SV, JAW, AP, KBM, TI, BFR, FKK, TV and SM critically revised the manuscript for intellectual content. All authors gave final approval of the version to be published. JJH is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wewer Albrechtsen, N.J., Hartmann, B., Veedfald, S. et al. Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels?. Diabetologia 57, 1919–1926 (2014). https://doi.org/10.1007/s00125-014-3283-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3283-z