
Abstract
Aims/hypothesis
The aim of this study was to quantify the relative contributions of increased insulin secretion rate (ISR) and decreased insulin clearance rate (ICR) in the compensatory hyperinsulinaemia characteristic of insulin-resistant individuals without diabetes.
Methods
Obese (BMI ≥30 kg/m2) individuals without diabetes (n = 91) were identified from a registry of volunteers. Volunteers underwent the following measurements: oral glucose tolerance; insulin resistance (steady-state plasma glucose [SSPG] concentration during the insulin suppression test [IST]); ISR (using the graded glucose infusion test [GGIT]); and ICR (using the IST and GGIT). Participants were stratified into tertiles based on SSPG concentration: SSPG-1(insulin-sensitive); SSPG-2 (intermediate); and SSPG-3 (insulin-resistant).
Results
There were no differences in BMI and waist circumference among the SSPG tertiles. Serum alanine aminotransferase concentrations were higher in the SSPG-2 and SSPG-3 groups compared with the SSPG-1 group (p = 0.02). Following an oral glucose challenge, there was a progressive increase in the total integrated insulin response from the most insulin-sensitive to the most insulin-resistant tertiles (p < 0.001). Following intravenous glucose, the SSPG-3 group had significantly greater integrated glucose (median [interquartile range], 32.9 [30.8–36.3] mmol/l × h) and insulin responses (1711 [1476–2223] mmol/l × h) compared with the SSPG-1 group (30.3 [28.8–32.9] mmol/l × h, p = 0.04, and 851 [600–1057] pmol/l × h, p < 0.001, respectively). Furthermore, only the SSPG-3 group had significant changes in both ISR and ICR (p < 0.001). In the SSPG-2 group, only the ICR was significantly decreased compared with the SSPG-1 group. Therefore, ICR progressively declined during the IST with increasing insulin resistance (SSPG-1, 0.48 [0.41–0.59]; SSPG-2, 0.43 [0.39–0.50]; SSPG-3, 0.34 [0.31–0.40]).
Conclusions/interpretation
While both increases in ISR and decreases in ICR compensate for insulin resistance, decreases in ICR may provide the first adaptation to decreased insulin sensitivity.
Similar content being viewed by others

Introduction
Currently, the pathogenesis of type 2 diabetes is thought to involve the failure of pancreatic beta cells to secrete sufficient insulin to satisfy demand in the context of obesity and/or increased insulin resistance [1].While this hypothesis is widely accepted, an increase in the insulin secretion rate (ISR) is not the only adaptation to insulin resistance. Decreases in the insulin clearance rate (ICR) also help to maintain hyperinsulinaemia in insulin-resistant states [2]. However, the relative roles of increases in ISR vs decreases in ICR in maintaining hyperinsulinaemia and glucose tolerance in insulin resistance are unclear.
The aim of this study was to better understand the physiological adaptations to insulin resistance prior to the development of type 2 diabetes in a sample of 91 obese individuals who have had direct measurements of insulin resistance using the insulin suppression test (IST). We compared glucose tolerance, ISR and ICR in equally obese individuals with varying degrees of insulin sensitivity.
Methods
The study population consisted of 91 individuals who had previously responded to an advertisement seeking volunteers for research studies on the role of insulin resistance in human disease. All individuals were apparently healthy without coronary, kidney or liver disease. The Stanford Institutional Review Board approved all studies and all participants provided informed consent.
We identified 152 individuals in our ongoing registry of participants who had undergone an OGTT, IST and graded glucose infusion test (GGIT). For the current study, we included individuals with obesity (BMI ≥30 kg/m2, n = 96). Five additional individuals were excluded for having 2 h glucose ≥11 mmol/l [3]. Therefore, 91 participants were included in the final sample.
The study population was stratified into tertiles based on their degree of insulin resistance as defined by their steady-state plasma glucose (SSPG) concentration during the IST as described below.
IST
Peripheral insulin resistance was directly measured with the modified version of the IST [4]. Measures of insulin action with the IST are highly correlated (r ≥ 0.87) with those obtained with the hyperinsulinaemic–euglycaemic clamp [5, 6]. In brief, after an overnight fast, an intravenous catheter was placed in each of the participants’ arms. One arm was used for the administration of a 180 min infusion of octreotide (0.27 mg m−2 min−1), insulin (32 mU m−2 min−1) and glucose (267 mg m−2 min−1); the other arm was used for collecting blood samples. Blood was drawn at 10 min intervals from 150 min to 180 min of the infusion to determine the SSPG and steady-state plasma insulin (SSPI) concentrations. Under these conditions SSPI concentration was comparable for all participants and the SSPG concentration provides a direct measure of the ability of insulin to mediate disposal of an infused glucose load; the higher the SSPG concentration, the greater the degree of insulin resistance. Based on the SSPG concentrations, the population was divided into tertiles:insulin-sensitive (SSPG-1); intermediate (SSPG-2); and insulin-resistant (SSPG-3).
OGTT
All participants underwent a 75 g OGTT in which plasma glucose and insulin concentrations were measured before and 30, 60, 120 and 180 min after the glucose challenge. The AUCs for glucose (G-AUC) and insulin (I-AUC) were calculated using the trapezoidal method.
GGIT
All participants received graded intravenous infusions of glucose at progressively increasing rates (1, 2, 3, 4, 6 and 8 mg kg−1 min−1) as previously described [2, 7,8,9]. Each glucose infusion rate was administered for a total of 40 min. Glucose, insulin and C-peptide concentrations were measured at 0, 30, 40, 70, 80, 110, 120, 150, 160, 190, 200, 230 and 240 min. The last two glucose and insulin values at the end of each 40 min infusion period (e.g. 30 and 40 min values) were averaged and used as the mean for that infusion.
Measurement of ISR
ISRs were derived by deconvolution covering 240 min and 13 sampling time points for peripheral plasma C-peptide concentrations, using a two-compartment model of C-peptide kinetics and standard variables for C-peptide clearance estimated for each participant based on body surface area and age [9]. Previously, we have also used 25 sampling time points [2, 7, 8]. In a subset of 14 individuals, we found no significant differences in ISR using 13 vs 25 sampling time points (S. H. Kim, unpublished data). For each participant, the mean ISR before and during the six glucose infusion periods was plotted against the corresponding mean glucose to construct a dose–response relationship. The beta cell sensitivity represented the slope of the line relating glucose and ISR.
Measurement of ICR
ICR was calculated during the IST and GGIT. During the IST, ICR was calculated by dividing the insulin infusion rate by the SSPI concentration [10, 11]. As octreotide is infused to inhibit endogenous insulin secretion, ICR during the IST represents exogenous ICR (ICRex). During the GGIT, ICR was calculated by using the following formula:
The AUCs for ISR and insulin were calculated using the trapezoidal method. V refers to volume of distribution of insulin, which was estimated as 0.14 l/kg [12]. I refers to insulin concentration at time (t) 0 min and 240 min.
Metabolic measurements
Glucose was determined by the oxidase method (Analyzer 2; Beckman, Brea, CA, USA). Insulin and C-peptide concentrations were measured at Washington University (St Louis, MO, USA) using radioimmunoassay (Millipore, St Charles, MO, USA). The inter- and intra-assay coefficient of variation ranged between 4.7% and 9.7% for insulin, and 5.2% and 10.9% for C-peptide. Creatinine and alanine aminotransferase (ALT) were measured in the clinical laboratory at Stanford Health Care (Stanford, CA, USA).
Statistical analysis
Statistical analysis was performed using SPSS (version 23 for Windows; SPSS, Armonk, NY, USA). Data are reported as mean ± SD or median (interquartile range). Non-parametric variables were log-transformed prior to analyses. Comparisons among groups were made using one-way ANOVA. A probability value of <0.05 was considered to be statistically significant in all analyses.
Results
Table 1 presents the demographic and clinical characteristics of the study population stratified into tertiles based on degree of insulin resistance (SSPG concentration). By selection, magnitude of insulin resistance increased progressively from the least (SSPG-1) to the most insulin-resistant tertile (SSPG-3). Despite the dramatic differences in SSPG concentration, the three groups were otherwise well matched and did not differ in age, sex distribution, race/ethnicity, BMI, waist circumference or fasting plasma glucose concentration. However, ALT concentrations were significantly higher and creatinine concentration was lower in the two most insulin-resistant groups (SSPG-2 and SSPG-3).
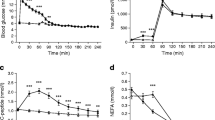
Plasma glucose and insulin concentrations following the oral glucose challenge are shown in Fig. 1. The total area under the glucose response curves (G-AUC) of the three groups are illustrated in Fig. 1a. The three groups were significantly different (p = 0.001), with the G-AUC of the most insulin-resistant tertile (SSPG-3) being significantly higher than the other two groups (p < 0.05). The insulin responses (I-AUC) of the three SSPG groups to oral glucose (Fig. 1b) separated more dramatically (p < 0.001) and I-AUC increased progressively from the most insulin-sensitive to the most insulin-resistant tertile.

(a) Glucose and (b) insulin concentrations during the OGTT are shown for the SSPG-1 (white circles), SSPG-2 (black triangles) and SSPG-3 (black squares) groups. G-AUC and I-AUC significantly differed among the three SSPG groups (p ≤ 0.001). The most insulin-resistant group (SSPG-3) had the highest G-AUC and I-AUC. Data are means ± SEM
Figure 2 presents the changes in plasma glucose and insulin concentrations and ISR in response to the graded glucose infusion in the three SSPG groups. The quantitative comparisons between the three groups are shown in Table 2. Plasma glucose concentrations increased modestly in all three groups (Fig. 2a), with the G-AUC being significant higher in the most insulin-resistant group (SSPG-3) compared with the most insulin-sensitive group (SSPG-1). The changes in insulin concentration and ISR (Fig. 2b, c) in response to the intravenous glucose infusion were more pronounced in the most insulin-resistant tertile (SSPG-3), and the AUCs were significantly greater in this group than in the other two groups.

(a) Glucose, (b) insulin and (c) ISR during the GGIT are shown for the SSPG-1 (white circles), SSPG-2 (black triangles) and SSPG-3 (black squares) groups. The G-AUC (p = 0.04), I-AUC (p < 0.001) and ISR-AUC (p < 0.001) differed among the groups, using ANOVA. The most insulin-resistant group (SSPG-3) had the highest G-AUC, I-AUC and ISR-AUC. In post hoc analysis, there were no significant differences between the SSPG-1 and SSPG-2 groups. Data are means ± SEM
The changes in ICR, as measured during the GGIT and IST, are illustrated in Fig. 3. During the GGIT, endogenous ICR (ICRen) differed among the three groups (p < 0.001). In contrast to insulin concentration and ISR, ICRen progressively decreased from the most insulin-sensitive to the most insulin-resistant tertiles (Fig. 3a). That is, ICRen was significantly lower in SSPG-2 vs SSPG-1 (p = 0.03) and SSPG-3 vs SSPG-2 (p = 0.003). The results were similar when ICRex was calculated during the IST (Fig. 3b). Thus, ICRex was highest in the SSPG-1 group, intermediate in the SSPG-2 group and lowest in the SSPG-3 group (all pairwise comparisons were significant, p < 0.05).

ICRen and ICRex are shown for the SSPG-1, SSPG-2 and SSPG-3 groups during the (a) GGIT and (b) IST. Both measures of ICR progressively declined from SSPG-1 to SSPG-3. All pairwise comparisons were significantly different (p < 0.05). Data are means ± SEM
Finally, the relationship between the plasma glucose concentration and ISR were compared among the three SSPG groups. As shown in Fig. 4, the curve in the most insulin-resistant group (SSPG-3) shifted upward and to the left. Thus, the individuals in the SSPG-3 group had significantly higher beta cell sensitivity to glucose (Table 2) and secreted more insulin at any given plasma glucose concentration compared with the other more insulin-sensitive groups.

The relationship between glucose and ISR is shown for the SSPG-1 (white circles), SSPG-2 (black triangles) and SSPG-3 (black squares) groups. The curve, representing beta cell sensitivity to glucose, was shifted up and to the left in the most insulin-resistant group (SSPG-3) compared with the other two groups (p = 0.01). Data are means ± SEM
Discussion
The aim of the current analysis was to understand how obese individuals adapt to the demands of insulin resistance prior to the development of type 2 diabetes. By evaluating equally obese individuals with varying degrees of insulin sensitivity, we were able to define the relative contributions of ISR and ICR in maintaining hyperinsulinaemia as the magnitude of insulin resistance increased. The most novel finding was seen in the group with intermediate insulin resistance (SSPG-2), who had a significant decrease in ICR but no change in ISR compared with the insulin-sensitive group (SSPG-1). The most insulin-resistant group (SSPG-3) had a significant decrease in ICR and an increase in ISR compared with the insulin-sensitive group. This suggests that the first compensation for insulin resistance is a decrease in ICR, which occurs prior to any significant increase in ISR.
Prior studies have shown significant differences in ISR and ICR in insulin-resistant vs insulin-sensitive groups [2, 13,14,15]. In the majority of these studies [13,14,15], degree of obesity and not direct measurement of insulin resistance was used to identify individuals as insulin-resistant vs insulin-sensitive. This is problematic as we have recently shown that obesity itself can increase ISR [16]. Furthermore, we have previously used the IST to identify non-obese insulin-sensitive and insulin-resistant individuals [2]. Similar to the comparisons between the SSPG-3 and SSPG-1 groups in the present study, we found significant increases in ISR and decreases in ICR in insulin-resistant individuals compared with insulin-sensitive individuals. However, since these comparisons were performed in dichotomous groups, insulin-resistant vs insulin-sensitive, the results did not permit an evaluation of the potential importance of differences in magnitude of insulin resistance.
By comparing the three SSPG groups, our study demonstrates the importance of ICR in adapting to the demands of insulin resistance, as it appears to occur prior to a significant increase in ISR. While we are unaware of other human studies demonstrating the early role of ICR in insulin resistance, a longitudinal study in dogs demonstrated the relevance of ICR in maintaining hyperinsulinaemia in insulin resistance [17]. The dogs were fat-fed for 12 weeks and had a significant increase in fasting hyperinsulinaemia and insulin resistance. ISR transiently increased at 6 weeks but returned to baseline at 12 weeks. At the end of the study, hyperinsulinaemia was maintained due to a significant decrease in ICR. The authors speculated that a decrease in insulin catabolism may help ‘rest’ the beta cells in the face of chronic insulin resistance. As our study was cross-sectional, we cannot comment on the dynamic changes in ISR and ICR as insulin resistance develops. However, it is clear that ICR plays an important and early role in maintaining hyperinsulinaemia in insulin resistance.
The mechanism for the decrease in ICR in insulin resistance remains unclear but could relate to an increase in hepatic lipid associated with insulin resistance [18]. The liver is the major site for insulin degradation [19] and hepatic steatosis has been shown to correlate negatively with ICR [20]. Consistent with this possibility are the findings of similarly elevated ALT concentrations in the SSPG-2 and SSPG-3 groups compared with the most insulin-sensitive group (SSPG-1).
Although ICR is important in maintaining hyperinsulinaemia in insulin resistance, the decrease in ICR is apparently not sufficient compensation in the most insulin-resistant tertile (SSPG-3), whose SSPG concentration was double that of the most insulin-sensitive tertile (SSPG-1). In these people, the hyperinsulinaemia is the result of both a decrease in ICR and an increase in ISR. Interestingly, the glucose concentration was also significantly, albeit modestly, increased in the SSPG-3 group compared with the SSPG-1 group. The modest increase in blood glucose is likely to have a direct effect on pancreatic beta cell insulin synthesis and secretion [21]. In support, beta cell sensitivity to glucose was significantly increased in the SSPG-3 group, as demonstrated by the upward shift in the ISR curve in Fig. 4.
Based upon our findings, we propose the following overall hypothesis to describe the adaptation to insulin resistance in obesity prior to the development of type 2 diabetes. A relative modest degree of insulin resistance is associated with an increase in hepatic fat content, leading to a decrease in ICR and, based on the findings shown in Fig. 1, an increase in plasma insulin concentrations sufficient to maintain euglycaemia. However, the decrease in ICR is not sufficient compensation in the most insulin-resistant tertile (SSPG-3); in these people, hyperinsulinaemia is the result of both a decrease in ICR and an increase in ISR.
There are limitations to our study. First, the data were cross-sectional, derived from prior experimental studies, and selected to include only obese individuals. Furthermore, despite the fact that overall (BMI) and abdominal (waist circumference) obesity of the three groups were essentially identical (Table 1), the analysis lacks direct measurements of regional fat distribution. Thus, the conclusion that the relationships among insulin resistance, plasma insulin concentration, ISR and ICR discerned in the analysis are independent of differences in adiposity should be tempered. Furthermore, we used a modified protocol for sampling blood (13 vs 25), which may have affected calculation of ISR and ICR. Nevertheless, the qualitative results for ICR were similar to calculations based on the IST. Finally, we did not measure hormones, in particular incretin hormones, which also play significant roles in postprandial glucose and insulin modulation [22]. Differences in incretin hormones may also explain the differences in plasma insulin concentration during the OGTT among the three groups compared with during the GGIT. Despite these shortcomings, we are unaware of published specific quantitative measurements of insulin resistance, ISR and ICR in obese, apparently healthy individuals over a wide range of insulin resistance. As such, our findings provide a coherent view demonstrating the relative changes in ICR and ISR with increases in insulin resistance. While changes in both ISR and ICR occur, decreases in ICR may provide the first adaptation to decreased insulin sensitivity.
Abbreviations
- ALT:
-
Alanine aminotransferase
- G-AUC:
-
Area under the curve for glucose
- GGIT:
-
Graded glucose infusion test
- I-AUC:
-
Area under the curve for insulin
- ICR:
-
Insulin clearance rate
- ICRen :
-
Endogenous insulin clearance rate
- ICRex :
-
Exogenous insulin clearance rate
- ISR:
-
Insulin secretion rate
- IST:
-
Insulin suppression test
- SSPG:
-
Steady-state plasma glucose
- SSPI:
-
Steady-state plasma insulin
References
Skyler JS, Bakris GL, Bonifacio E et al (2017) Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 66:241–255
Jones CN, Pei D, Staris P, Polonsky KS, Chen YD, Reaven GM (1997) Alterations in the glucose-stimulated insulin secretory dose-response curve and in insulin clearance in nondiabetic insulin-resistant individuals. J Clin Endocrinol Metab 82:1834–1838
American Diabetes Association (2011) Diagnosis and classification of diabetes mellitus. Diabetes Care 34(Suppl 1):S62–S69
Pei D, Jones CN, Bhargava R, Chen YD, Reaven GM (1994) Evaluation of octreotide to assess insulin-mediated glucose disposal by the insulin suppression test. Diabetologia 37:843–845
Greenfield MS, Doberne L, Kraemer F, Tobey T, Reaven G (1981) Assessment of insulin resistance with the insulin suppression test and the euglycemic clamp. Diabetes 30:387–392
Knowles JW, Assimes TL, Tssao PS et al (2013) Measurement of insulin-mediated glucose uptake: direct comparison of the modified insulin suppression test and the euglycemic, hyperinsulinemic clamp. Metabolism 62:548–553
Jones CN, Abbasi F, Carantoni M, Polonsky KS, Reaven GM (2000) Roles of insulin resistance and obesity in regulation of plasma insulin concentrations. Am J Physiol Endocrinol Metab 278:E501–E508
Kim SH, Abbasi F, Chu JW et al (2005) Rosiglitazone reduces glucose-stimulated insulin secretion rate and increases insulin clearance in nondiabetic, insulin-resistant individuals. Diabetes 54:2447–2452
Van Cauter E, Mestrez F, Sturis J, Polonsky KS (1992) Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes 41:368–377
Kim MK, Reaven GM, Chen YI, Kim E, Kim SH (2015) Hyperinsulinemia in individuals with obesity: role of insulin clearance. Obesity 23:2430–2434
Kim S, Liu A, Ariel D et al (2014) Effect of salsalate on insulin action, secretion, and clearance in nondiabetic, insulin-resistant individuals: a randomized, placebo-controlled study. Diabetes Care 37:1–7
Polidori DC, Bergman RN, Chung ST, Sumner AE (2016) Hepatic and extrahepatic insulin clearance are differentially regulated: results from a novel model-based analysis of intravenous glucose tolerance data. Diabetes 65:1556–1564
Polonsky KS, Given BD, Hirsch L et al (1988) Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 81:435–441
Bonora E, Zavaroni I, Bruschi F et al (1984) Peripheral hyperinsulinemia of simple obesity: pancreatic hypersecretion or impaired insulin metabolism? J Clin Endocrinol Metab 59:1121–1127
Meistas MT, Margolis S, Kowarski AA (1983) Hyperinsulinemia of obesity is due to decreased clearance of insulin. Am J Phys 245:E155–E159
Kim MK, Reaven GM, Kim SH (2017) Dissecting the relationship between obesity and hyperinsulinemia: role of insulin secretion and insulin clearance. Obesity 25:378–383
Kim SP, Ellmerer M, Kirkman EL, Bergman RN (2007) Beta-cell “rest” accompanies reduced first-pass hepatic insulin extraction in the insulin-resistant, fat-fed canine model. Am J Physiol Endocrinol Metab 292:E1581–E1589
Choudhury J, Sanyal AJ (2004) Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin Liver Dis 8:575–594
Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19:608–624
Kotronen A, Vehkavaara S, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H (2007) Effect of liver fat on insulin clearance. Am J Physiol Endocrinol Metab 293:E1709–E1715
Cavaghan MK, Ehrmann DA, Polonsky KS (2000) Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J Clin Invest 106:329–333
Kim W, Egan JM (2008) The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev 60:470–512
Acknowledgements
The authors would like to thank the volunteers and the staff and nurses in the Stanford Clinical and Translational Research Unit for their invaluable assistance with our metabolic studies. Some of the data in this manuscript were presented as an abstract at the ADA 77th Scientific Sessions in 2017.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The datasets analysed during the current study are not publicly available for reasons of privacy. Anonymous data is available from the corresponding author on reasonable request.
Funding
This study was supported in part by an NIH/NCRR CTSA award number UL1 RR025744 and a gift from the Bose Family Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
SJ, CJ, GR and SK contributed to the study design. GR and SK contributed to data acquisition. SJ wrote the manuscript and researched data. CJ contributed to the discussion. GR reviewed/edited the manuscript. SK researched data and reviewed/edited the manuscript. All authors drafted or revised the article and approved the final version of the manuscript. SK is the guarantor of this work, had full access to all the data in the study and takes responsibility for the integrity of the data and accuracy of the data analysis.
Rights and permissions
About this article

Cite this article
Jung, SH., Jung, CH., Reaven, G.M. et al. Adapting to insulin resistance in obesity: role of insulin secretion and clearance. Diabetologia 61, 681–687 (2018). https://doi.org/10.1007/s00125-017-4511-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4511-0