
Abstract
O-linked β-N-actylglucosamine (O-GlcNAc) is a carbohydrate post-translational modification on hydroxyl groups of serine and/or threonine residues of cytosolic and nuclear proteins. Analogous to phosphorylation, O-GlcNAcylation plays crucial regulatory roles in a variety of cellular processes. O-GlcNAc was termed a nutritional sensor, as global levels of the modification are elevated in response to increased glucose and glutamine flux into the hexosamine biosynthetic pathway. A unique feature of cancer cell energy metabolism is a shift from oxidative phosphorylation to the less efficient glycolytic pathway (Warburg effect), necessitating greatly increased glucose uptake. Additionally, to help meet increased biosynthetic demands, cancer cells also up-regulate glutamine uptake. This led us to hypothesize that the universal feature of increased glucose and glutamine uptake by cancer cells might be linked to increased O-GlcNAc levels. Indeed, recent work in many different cancer types now indicates that hyper-O-GlcNAcylation is a general feature of cancer and contributes to transformed phenotypes. In this review, we describe known/potential links between hyper-O-GlcNAcylation and specific hallmarks of cancer, including cancer cell proliferation, survival, cell stresses, invasion and metastasis, aneuploidy, and energy metabolism. We also discuss inhibition of hyper-O-GlcNAcylation as a potential novel therapeutic target for cancer treatment.
Similar content being viewed by others


Introduction
O-linked β-N-actylglucosamine (O-GlcNAc), which was discovered in the early 1980s (Torres and Hart 1984), is the covalent addition of an N-actylglucosamine (GlcNAc) sugar moiety to hydroxyl groups of serine and/or threonine residues of cytosolic and nuclear proteins. Modification of proteins by O-GlcNAc (O-GlcNAcylation) is unique among a myriad of carbohydrate post-translational modifications, as it is almost exclusively cytosolic and nuclear and generally not further modified to more complex glycans. The O-GlcNAc transferase (OGT) catalyzes enzymatic addition of O-GlcNAc by transfer of the GlcNAc moiety from the high-energy donor substrate UDP-GlcNAc, an end-product of the hexosamine biosynthetic pathway (HBP) (Fig. 1). O-GlcNAc is present in all metazoans studied thus far and ubiquitous in all cells and tissues. O-GlcNAc removal is catalyzed by O-GlcNAcase (OGA). The balance of activity of OGT and OGA determines the cycling rate of O-GlcNAc on proteins. O-GlcNAcylation, analogous to phosphorylation, plays crucial regulatory roles on many proteins in signal transduction that impacts a variety of cellular processes including cell cycle progression, transcription, cellular stress responses, and epigenetic control of gene expression (Hart et al. 2007; Hart 1997; Wells et al. 2001; Zachara and Hart 2004; Haltiwanger et al. 1992).

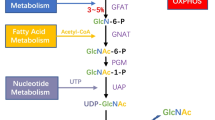
Cancer cell metabolic changes linked to hyper-O-GlcNAcylation. The hexosamine biosynthetic pathway (HBP) outlined in orange boxes integrates metabolic intermediates to generate the end-product UDP-GlcNAc. Glucose is transported into cells by glucose transporters such as Glut1 and then first phosphorylated by hexokinase to generate glucose-6-phosphate. Glucose-6-phosphate can be shunted into the PPP which produces nucleotides and NAPDH, or converted into fructose-6-phosphate. While most fructose-6-phosphate continues through glycolysis to produce pyruvate, some is directed into the HBP. GFAT, the HBP rate-limiting enzyme, irreversibly transfers the amino group from glutamine to fructose-6-phosphate, generating glucosamine-6-phosphate and glutamate. Glucosamine-6-phosphate is ultimately converted to UDP-GlcNAc, which is used by OGT to attach O-GlcNAc to hydroxyl groups of serine and/or threonine residues of cytosolic and nuclear proteins. O-GlcNAc is removed by OGA. Cancer cell metabolic changes including increased glucose uptake (due to “Warburg effect”) and increased glutamine uptake (along with elevated UTP and acetyl-CoA production) cooperate to maximize flux through the HBP. Oncogenes such as HIF1α, Kras, and c-Myc regulate cancer cell shifts to aerobic glycolysis and glutaminolysis, including upregulation of glucose and glutamine transporters and increased expression of GFAT. Additionally, the level of OGT is increased and the level of OGA is decreased. In sum, cancer cell metabolic reprogramming leads to increased HBP flux, elevated UDP-GlcNAc, and ultimately hyper-O-GlcNAcylation. Proteins and metabolic intermediates in red are increased in cancer cells. G6P: Glucose-6-phosphate; F6P: fructose-6-phosphate; FBP fructose 1,6-bisphosphate, PEP phosphoenolpyruvate, GFAT glutamine: fructose-6-phosphate amidotransferase, MCT4 monocarboxylate transporter, OAA oxaloacetate, PFK1 phosphofructokinase 1, PKM2 pyruvate kinase M2 (color figure online)
Recently, we and other groups have shown that elevated O-GlcNAcylation (hyper-O-GlcNAcylation) occurs in human malignancies including breast (Caldwell et al. 2010; Gu et al. 2010), prostate (Lynch et al. 2012), lung (Mi et al. 2011), colorectal (Mi et al. 2011; Yehezkel et al. 2012), liver (Zhu et al. 2012), pancreatic cancer (Ma et al. 2013), and non-solid cancers such as chronic lymphocytic leukemia (Shi et al. 2010). Hyper-O-GlcNAcylation seems to be a general feature of cancer cells. Furthermore, reducing hyper-O-GlcNAcylation inhibits cancer phenotypes and blocks tumor growth in a variety of cancer models (Ma et al. 2013; Lynch et al. 2012; Caldwell et al. 2010), indicating that hyper-O-GlcNAcylation contributes to transformation.
A number of hallmarks of cancer have been proposed as a way to categorize the complex cellular changes that occur in transformation (Hanahan and Weinberg 2011; Luo et al. 2009). Owing to the plethora of molecular changes in cancer and widespread diverse regulatory roles of O-GlcNAc, there are likely numerous and complex links between cancer cell hyper-O-GlcNAcylation and underlying oncogenic cellular processes. In this review, we discuss how metabolic reprogramming in cancer is linked to aberrant increased O-GlcNAcylation. We then describe emerging evidence for specific hyper-O-GlcNAcylation events linked to pro-oncogenic signaling in the context of various hallmarks of cancer. Finally, we discuss the clinical potential of therapeutically targeting hyper-O-GlcNAcylation in cancer.
Links between cancer cell metabolic reprogramming and hyper-O-GlcNAcylation
One of the remarkable features of cancer cells is aerobic glycolysis, a phenomenon also known as the “Warburg effect” (Warburg et al. 1927; Warburg 1956a, b), in which cancer cells rely preferentially on glycolysis instead of oxidative phosphorylation as the main energy source even in the presence of high oxygen tension. Glycolysis is much less efficient than oxidative phosphorylation in producing ATP, necessitating greatly increased cancer cell glucose uptake. It may appear paradoxical that a highly proliferating cancer cell with increased energy needs would shift metabolically to glycolysis. However, it was proposed that such metabolic reprogramming may in part serve to balance bio-energetic and biosynthetic needs of rapidly dividing cancer cells, as excess glucose taken up may also serve as precursors for increased cancer cell needs for biosynthesis of molecules such as nucleotides and lipids (Fig. 1) (Kroemer and Pouyssegur 2008; Vander Heiden et al. 2009; DeBerardinis et al. 2008). The switch to glycolysis in cancer cells is driven in part by tumor cell hypoxia, oncogenes (e.g., Ras, Myc), and mutant tumor suppressors (e.g., TP53), which activate HIF-1α, PI3K/Akt/mTOR, and c-Myc pathways to promote Warburg-like tumor metabolism (DeBerardinis et al. 2008). One main consequence is up-regulation of glucose transporters, especially GLUT1 [e.g., by HIF-1α and c-Myc (Osthus et al. 2000)], which significantly increases glucose import into tumor cells. Due to this property of cancers, glucose analogs such as 18F-fluoro-2-deoxy-glucose (FDG) are widely used in PET/CT imaging of tumors to diagnose and evaluate therapeutic responses in cancer patients (Vander Heiden et al. 2009). The abundance of glucose in cancer cytoplasm not only contributes to increased glycolytic flux, but also increases flux into metabolic branch pathways of glycolysis. For example, increased pentose phosphate pathway (PPP) flux contributing to increased nucleotide synthesis (e.g., UTP) is observed in cancers. Additionally, about 2–5 % of glucose entering a cell fluxes through the HBP. Thus, increased cancer cell glucose uptake may drive increased HBP flux. Indeed, certain oncogenes such as Kras not only up-regulate levels of glucose transporters and glycolytic enzymes, but up-regulate glutamine: fructose-6-phosphate amidotransferase 1 (GFAT1), the rate-limiting enzyme in the HBP (Ying et al. 2012). Thus, both excess glucose uptake and up-regulation of HBP enzymes likely drive increased HBP flux in cancer.
Cancer cells are also addicted to glutamine (Yuneva et al. 2007). Cancer cells consume glutamine at high rates in vivo and compared to non-transformed cells require high concentrations of glutamine to survive and proliferate (DeBerardinis 2008). Oncogenes can up-regulate glutamine uptake. For example, c-Myc transcriptionally up-regulates glutamine transporter expression (Wise et al. 2008; Yuneva et al. 2007; Gao et al. 2009).
One fate of glutamine in cancer cells is conversion to oxaloacetate (OAA) via glutamate and α-ketoglutarate (α-KG), in a process termed anaplerosis, to resupply the mitochondrial TCA intermediate OAA. Cancer cells employ a “broken” or “truncated” TCA cycle that shunts citrate into production of acetyl-CoA for increased needs of fatty acid synthesis (DeBerardinis 2008; Moreno-Sanchez et al. 2007). Citrate can be regenerated from OAA. The “lost” OAA can be replenished during anaplerosis either by pyruvate via glycolysis or by glutamine. Glutamine may also undergo partial oxidization to generate lactate and NADPH (glutaminolysis) to produce energy in a manner analogous to aerobic glycolysis (DeBerardinis et al. 2008). Additionally, glutamine is the donor substrate in the conversion of fructose-6-phosphate to glucosamine-6-phosphate by GFAT in the HBP. Thus, under conditions in which GFAT is not limiting, excess glutamine uptake in cancer cells could contribute to increased flux through the HBP, ultimately contributing to increased levels of the HBP end-product UDP-GlcNAc. Indeed, in vivo glutamine administration itself was shown to increase HBP flux (Liu et al. 2007; Singleton and Wischmeyer 2008). Such metabolic reprogramming in cancer cells meets energetic demands for rapid cell proliferation and supplies increased intermediates for cancer cell biosynthesis. Consequently, cancer cell metabolic changes including increased glucose uptake due to the “Warburg effect”, and increased glutamine uptake likely cooperate to drive increased HBP flux. In support of this, we find that the end products of the HBP (including UDP-GlcNAc) are elevated in pancreatic cancer cells (Ma et al. 2013).
The HBP end-product UDP-GlcNAc is the donor substrate used by OGT in enzymatic addition of O-GlcNAc. Increased HBP flux was linked to increased levels of O-GlcNAc (Wells et al. 2003). This led us to hypothesize that cancer metabolism may drive increased levels of O-GlcNAc. Indeed, hyper-O-GlcNAcylation is observed in all cancer types examined thus far (Caldwell et al. 2010; Lynch et al. 2012; Ma et al. 2013; Mi et al. 2011; Shi et al. 2010; Zhu et al. 2012). Additionally, loss of the p53 tumor suppressor in mouse embryonic fibroblasts (MEFs) increases the rate of aerobic glycolysis, the expression of GLUT3 (Kawauchi et al. 2008), increases HBP flux and leads to elevation of O-GlcNAcylation (Kawauchi et al. 2009). Thus, it appears that increased HBP flux and elevated UDP-GlcNAc is a general feature of cancer cells that contributes to hyper-O-GlcNAcylation.
Altered expression levels of enzymes involved in O-GlcNAc cycling may contribute to cancer cell hyper-O-GlcNAcylation
Glutamine: fructose-6-phosphate amidotransferase, the rate-limiting enzyme in the HBP, irreversibly transfers the amino group from glutamine to fructose-6-phosphate, generating glucosamine-6-phosphate and glutamate. Thus, deregulation of GFAT would likely influence HBP flux. In breast cancer MDA-MB-468 cells, EGF was shown to increase GFAT mRNA (Roos et al. 1996). Moreover, tumor hypoxia and HIF-1α transcriptionally induce expression of GFAT (Manzari et al. 2007). Conversely, down-regulation of oncogenic Kras or c-Myc in pancreatic cancer decreases the expression of GFAT (Ying et al. 2012). Thus, in addition to increased glucose and glucosamine uptake in cancer, deregulation of GFAT by oncogenic factors may also contribute to up-regulation of HBP flux.
Accumulating evidence indicates that OGT and OGA expression are deregulated in various cancers. We have shown that OGT is overexpressed in breast (Caldwell et al. 2010), prostate (Lynch et al. 2012), and pancreatic cancer cells (Ma et al. 2013), compared to non-transformed immortalized counterpart cells, while OGA levels are reduced in cancer cells. The same pattern of elevated OGT and reduced OGA is also observed in lung and colon cancer tissue, compared with the corresponding adjacent tissues (Mi et al. 2011). Thus, the pattern of increased OGT (catalyzing addition of O-GlcNAc) and reduced OGA (catalyzing removal of O-GlcNAc) in cancers likely contribute to driving hyper-O-GlcNAcylation. We further examined OGT and OGA gene expression changes using the data-mining platform Oncomine™ (Rhodes et al. 2004, 2007). Compared to normal cells, OGT transcripts are increased in a variety of cancers including prostate cancer, leukemia, colorectal cancer, bladder cancer, sarcoma and leukemia, while OGA transcripts are decreased in lymphoma, brain and ovarian cancer, and leukemia (http://www.oncomine.org). Although no mutations in OGT and OGA have been reported in human cancers, in a case of fibroblastic sarcoma, a genetic breakpoint was identified in TGFBR3 in 1p22 and in or near MGEA5 encoding OGA in 10q24 (Hallor et al. 2009), which could in part explain reduced expression of OGA in this case. Recent crystal structure of human OGT with a peptide substrate suggests that the limiting factor for O-GlcNAcylation is not polypeptides but UDP-GlcNAc (Lazarus et al. 2011). Increasing intracellular concentrations of UDP-GlcNAc also results in increased global O-GlcNAcylation by enhancing the activity of OGT (Kreppel and Hart 1999). Therefore, in addition to increased flux through the HBP, increased expression of OGT and reduced expression of OGA in cancer cells likely contributes to hyper-O-GlcNAcylation.
Intriguingly, there appears to be negative feedback mechanisms which cells use to attempt to buffer large changes in levels of O-GlcNAc. For example, elevation of O-GlcNAc in 3T3-L1 adipocyte cells by pharmacological inhibition of OGA (Slawson et al. 2005), leads to decreased OGT expression and increased OGA expression. Conversely, pharmacologically lowering O-GlcNAc levels in 3T3-L1 and HeLa cells results in higher OGT expression and lower OGA expression (Slawson et al. 2005). These changes in patterns of OGT and OGA expression are consistent with feedback signals to “normalize” or dampen abnormal increases or decreases in O-GlcNAc levels, although the mechanisms underlying such a feedback mechanism are not known. Despite these negative feedback mechanisms which attempt to “normalize” changing O-GlcNAc levels, cancer cells maintain a strikingly elevated level of O-GlcNAcylation (hyper-O-GlcNAcylation), elevated OGT, and decreased OGA compared to non-transformed cells. Thus, to some degree, it appears that cancers cells bypass this negative feedback mechanism which attempts to “normalize” O-GlcNAc levels. This hyper-O-GlcNAcylation contributes to transformed phenotypes, as reduction of hyper-O-GlcNAcylation in breast, prostate, and pancreatic cancer models inhibits cancer cell proliferation and tumor growth in vitro and in vivo. Cancer cells appear to be selectively addicted to this hyper-O-GlcNAcylation, as we observed that knocking down levels of OGT to the same extent in breast and pancreatic transformed versus non-transformed counterpart cells selectively causes death of transformed cells (Caldwell et al. 2010; Ma et al. 2013). As cancer cells appear to become dependent on this state of hyper-O-GlcNAcylation, strategies to target inhibition of OGT for reduction of cancer hyper-O-GlcNAcylation may be an effective anti-cancer approach, as a therapeutic window in which growth/survival of cancer cells may selectively be inhibited appears to exist. The potential mechanisms through which hyper-O-GlcNAcylation supports cancer phenotypes will be discussed below in the context of various hallmarks of cancer.
Hyper-O-GlcNAcylation and potential links to excessive cancer cell proliferation
Sustaining proliferative signaling and evading growth suppressors are two hallmarks acquired by cancer cells to achieve infinite replicative potential (Hanahan and Weinberg 2011) (Fig. 2). Oncogenic changes often promote cell cycle progression and bypass cell cycle checkpoints. FoxM1 acts as a key positive regulator of cell cycle progression by up-regulating transcription of genes involved in the G1/S and G2/M transition and its overexpression is linked to oncogenesis (Myatt and Lam 2007). O-GlcNAcylation is implicated in the stability of FoxM1. Reducing breast cancer cell hyper-O-GlcNAcylation decreases levels of FoxM1 and its target genes and increases cyclin-dependent kinase cell cycle inhibitor p27Kip1. Consistent with this, reducing hyper-O-GlcNAcylation decreases cell-cycle progression in breast and prostate cancer (Caldwell et al. 2010; Lynch et al. 2012). We have also shown that reducing hyper-O-GlcNAcylation inhibits the expression of Cyclin D1, which is a positive regulator of the G1/S transition (Ma et al. 2013). Thus, cancer cell hyper-O-GlcNAcylation appears in part to contribute to excessive growth through up-regulation of key proteins that drive cell cycle progression and down-regulation of cell cycle inhibitory proteins such as p27Kip1.

O-GlcNAc and hallmarks of cancer. Known and possible links between elevated O-GlcNAcylation (hyper-O-GlcNAcylation) and hallmarks of cancer are depicted. Hallmarks of cancer include excessive cell proliferation (sustaining proliferative signaling and evading growth suppressors), deregulated cellular energetics, sustained angiogenesis, tissue invasion and metastasis, resistance of cell death, replicative immortality, oxidative stress, proteotoxic stress, and metabolic stress. Proteins in red are increased in the level of expression, stability, or activity by hyper-O-GlcNAcylation and those in green are decreased in their expression level or activity. “G” in green oval denotes O-GlcNAcylation. GFAT glutamine: fructose-6-phosphate amidotransferase, GSH reduced glutathione, HSF1 heat shock factor 1, MMPs matrix metalloproteinases, PFK1 phosphofructokinase 1, PK pyruvate kinase, PPP pentose phosphate pathway (color figure online)
Hyper-O-GlcNAcylation promotes cancer cell survival
Cancer cells are subject to a number of physiologic stresses which may trigger cell death in non-transformed cells, such as nutrient stress, proteotoxic stress, oxidative stress, and hypoxia. As a result, cancer cells evolve mechanisms to resist cell death, which is one of the hallmarks of cancer (Hanahan and Weinberg 2011). Resistance to cell death in cancer cells may involve alteration of several cell death mechanisms. Most notable is that cancer cells elude the barrier to unrestricted cell growth imposed by programmed cell death by evolving anti-apoptotic mechanisms (Hanahan and Weinberg 2011).
Conditional knockout of OGT in mice results in loss of O-GlcNAc and T cell apoptosis (O’Donnell et al. 2004), suggesting hyper-O-GlcNAcylation in cancer may play an anti-apoptotic role. Indeed, we have found that reducing hyper-O-GlcNAcylation in pancreatic ductal adenocarcinoma (PDAC) cell lines MiaPaCa-2 and PANC-1 decreases the expression of the anti-apoptotic protein Bcl-xL, and induces pro-apoptotic cleavage of caspases-9 and 3, suggesting that reducing hyper-O-GlcNAcation triggered the intrinsic apoptotic pathway, which was confirmed by Annexin V/PI staining (Ma et al. 2013). Conversely, increasing O-GlcNAc in pancreatic cancer BxPC-3 cells protects against suspension-induced apoptosis (Ma et al. 2013). Up-regulated/constitutively activated NF-κB contributes to transformation in many cancers, in part through anti-apoptotic influences (Perkins 2012). We have shown that the NF-κB p65 subunit and upstream kinases IKKα/IKKβ are hyper-O-GlcNAcylated in pancreatic cancer (PDAC) cell lines. Reducing hyper-O-GlcNAcylation decreases PDAC cell p65 activating phosphorylation (S536), nuclear translocation, NF-κB transcriptional activity, and target gene expression. Conversely, mimicking PDAC hyper-O-GlcNAcylation through pharmacological elevation of O-GlcNAcylation increases IKKα and p65 O-GlcNAcylation, accompanied by activation of NF-κB signaling (Ma et al. 2013). Therefore, cancer cell hyper-O-GlcNAcyaltion appears to be anti-apoptotic, possibly in part through activation of NF-κB signaling.
Cancer hyper-O-GlcNAcylation may contribute to stress resistance
Cancer cells are subject to various stresses, such as oxidative stress, ER stress, and proteotoxic stress, which were proposed to represent additional hallmarks of cancer (Luo et al. 2009). Mechanisms developed by cancer cells to cope with these stresses are essential to cancer cell survival. Emerging evidence indicates that hyper-O-GlcNAcylation may contribute to pro-oncogenic cancer cell survival by combating such stresses.
Reactive oxygen species (ROS) can be generated either endogenously by normal aerobic metabolism or derived exogenously from the extracellular milieu (Martindale and Holbrook 2002). Excessive ROS production exceeding capacity of cellular antioxidant defenses leads to oxidative stress, which can lead to severe damage of nuclear or mitochondrial DNA, intracellular lipids, and proteins. The relationship between oxidative stress and cancer is complex. Cancer cells experience increased oxidative stress due to many reasons including rapid growth, impaired mitochondrial function, and a reactive stroma (Benz and Yau 2008). Oxidative stress may contribute to oncogenesis (Reuter et al. 2010). Conversely, excessive oxidative stress in cancer cells must be combated to avoid cell death (Martindale and Holbrook 2002). One potential connection between cancer cell protection against oxidative stress and hyper-O-GlcNAcylation is PFK1, the rate-limiting enzyme in glycolysis. Reduced glutathione (GSH) is critical in combating oxidative stress. The co-factor NADPH is required to maintain pools of reduced GSH. O-GlcNAcylation of PFK1 inhibits its activity in cancer cells (Yi et al. 2012), decreasing rates of flux through glycolysis and thus increasing shunt of fructose-6-phosphate into the PPP. A consequence of increased PPP flux is increased generation of NAPDH. Thus, hyper-O-GlcNAcylation in cancer cells would likely increase PPP flux and NADPH production contributing to maintaining a pool of reduced glutathione (GSH) to combat ROS-induced cell death (Yi et al. 2012). It has also been shown that increased O-GlcNAcylation inhibits hydrogen peroxide-induced ROS production in cardiomyocytes, although the underlying mechanism is not clear (Ngoh et al. 2011). Finally, a FOXO4-dependent oxidative stress response has been reported. O-GlcNcylation of FOXO4 increases its transcriptional activity, suggesting that hyper-O-GlcNAcylation may contribute anti-oxidative stress influence through FOXO4 (Ho et al. 2010). However, whether this FOXO4-dependent anti-oxidative mechanism is operative in cancer cells was not examined.
Cellular protein homeostasis, or proteostasis, refers to the cells capacity to maintain proper protein folding, trafficking, and avoidance of toxic protein aggregation (Balch et al. 2008). Proteostasis is maintained, under stress conditions, by a number of cellular response mechanisms including the heat shock response (HSR). HSR in mammals is mediated by six groups of closely related proteins: HSP100, HSP90, HSP70, HSP60, HSP40, and small HSPs, which are transcriptionally regulated by heat shock transcription factor (HSF1) (Balch et al. 2008; Dai et al. 2012). HSR is frequently activated/up-regulated in cancer cells and helps counter excessive proteotoxic stress due to various cancer associated states including increased gene copy number linked to aneuploidy and increased protein translation, misfolding, and/or trafficking (Luo et al. 2009). Indeed, targeting the HSR is anti-tumorigenic. For instance, small molecule inhibition or RNA interfering-mediated knockdown of HSP90 or its cognate partner HSP70 suppresses cancer cell growth and triggers apoptosis in multiple cancer types (Neckers and Workman 2012; Xia et al. 2012). In addition, loss of the master regulator HSF1 in HSR diminishes tumor growth either induced by Ras or p53 oncogenic mutations in mice or in various cancer cell lines (Dai et al. 2007; Dudeja et al. 2011). Several lines of evidence suggest that hyper-O-GlcNAcylation may potentially support cancer cell growth through increasing the levels and/or activity of HSPs. For example, cell stress-induced elevation of O-GlcNAc is protective against cell damage at least in part through induction of increased HSP levels, including HSP70 and HSP90 (Zachara et al. 2004). Lowering O-GlcNAcylation in MEFs decreases the expression of HSP70 and HSP40 (Zachara et al. 2004; Kazemi et al. 2010). Moreover, increasing HBP pathway flux by glutamine treatment to elevate O-GlcNAc enhances expression of HSF1 and HSP70 in a septic mouse model and in isolated rat cardiomyocytes (Singleton and Wischmeyer 2008; Gong and Jing 2011). HSP70 is modified by O-GlcNAc (Walgren et al. 2003). HSP70 also appears to display lectin-like binding activity specifically toward O-GlcNAc (Guinez et al. 2004). Furthermore, we and other groups have recently shown that O-GlcNAc on proteins such as tau, TAK1-binding protein, α-Synuclein, and PFK1 inhibits oligomerazation or aggregation in the context of disease states (Yuzwa et al. 2012; Yi et al. 2012; Marotta et al. 2012). Taken together, these results suggest that hyper-O-GlcNAcyaltion in cancer cells may reduce proteotoxic stress through several mechanisms, including transcriptional induction of HSP proteins, stabilization of HSPs through direct O-GlcNAc modification, or recruitment of HSPs to O-GlcNAc-modified targets through the lectin-like activity seen in the case of HSP70. Additionally, elevated cancer O-GlcNAcylation may contribute to reducing toxic protein aggregation.
Hyper-O-GlcNAcylation may contribute to cancer angiogenesis
Induction of angiogenesis during tumorigenesis is another hallmark of cancer (Hanahan and Weinberg 2011). Angiogenesis is not only essential for delivery of oxygen and nutrients to the interior of a tumor, but also contributes to allowing cancer cells to invade surrounding tissue and disseminate to distant organs (Hanahan and Folkman 1996). A variety of pro-angiogenic factors were linked to proliferation and differentiation of endothelial cells, such as vascular endothelial growth factors (VEGFs) and fibroblast growth factors (FGFs) (Baeriswyl and Christofori 2009). VEGF released by tumor cells stimulates the sprouting and proliferation of endothelial cells in growing tumor blood vessels by binding to VEGF receptors. FGFs have also been shown to sustain tumor angiogenesis by binding and activating their receptors on endothelial cells (Baeriswyl and Christofori 2009). Recent evidence indicates that tumor angiogenesis may in part be supported by hyper-O-GlcNAcylation. Reducing hyper-O-GlcNAcylation in the prostate cancer cell line PC-3ML by knocking down OGT inhibits the expression of VEGF and in vitro angiogenesis (Lynch et al. 2012). Furthermore, O-GlcNAc is required in FGF signal transduction in Drosophila (Mariappa et al. 2011). In order to sustain angiogenesis, the tumor stroma requires continuous remodeling, a process in which matrix metalloproteinases (MMPs) like MMP-2, MMP-9 are vital in proteolytic degradation of extracellular matrix (ECM) (Weis and Cheresh 2011). MMP-9 has also been shown to be important in liberating matrix-bound VEGF-A in mouse models (Bergers et al. 2000). Reducing hyper-O-GlcNAcylation in prostate cancer PC-3ML cells and in liver cancer HepG2 cells suppresses the expression of MMP-2 and -9 (Lynch et al. 2012; Zhu et al. 2012). Conversely, elevating O-GlcNAcylation using the OGA inhibitor thiamet-G or knockdown of OGA enhances the activity of MMP-2 and -9 in chondrocytes and increases the expression of MMP-1, -2, and -3 (Andres-Bergos et al. 2012; Zhu et al. 2012). Therefore, hyper-O-GlcNAcylation in tumors may contribute to angiogenesis through up-regulation of VEGF, MMPs, and FGF signaling.
Hyper-O-GlcNAcylation may contribute to cancer cell invasion and metastasis
Tumor metastasis (not the primary tumor) accounts for over 90 % of cancer mortality (Gupta and Massague 2006). From a clinical/translational point of view, the ability of tumor cells to invade and metastasize to distant organs is a critical hallmark of cancer (Hanahan and Weinberg 2011). Emerging evidence suggests that hyper-O-GlcNAcylation in cancers may be involved in tumor invasion and metastasis. Whereas increasing hyper-O-GlcNAcylation enhances the migration/invasion of breast and liver cancer cells, lowering hyper-O-GlcNAcylation by knockdown of OGT inhibits tumor invasion and metastasis in vivo and in vitro in breast and prostate cancer cells (Caldwell et al. 2010; Gu et al. 2010; Park et al. 2010a; Lynch et al. 2012). The mechanisms of how hyper-O-GlcNAcylation may regulate tumor invasion and metastasis are only beginning to be understood. Different cancer types may utilize distinct and/or overlapping tumor invasion/metastasis mechanisms (Friedl and Wolf 2003). One mechanism that contributes to metastasis is “epithelial to mesenchymal transition” (EMT) (Thiery et al. 2009; Thiery and Sleeman 2006). EMT involves loss of epithelial markers like E-cadherin, and gain of mesenchymal markers such as Vimentin and N-cadherin, which is coordinated by a number of transcription factors, including Zeb1, Zeb2, Twist, Slug, and Snail (Thiery et al. 2009; Valastyan and Weinberg 2011). We and others have found that reducing hyper-O-GlcNAcylation in cancers increases the expression of the epithelial marker E-cadherin and decreases expression of the mesenchymal marker Vimentin, while elevating O-GlcNAc decreases expression of the epithelial marker E-cadherin in breast and liver cancer (Gu et al. 2010; Ma et al. 2013; Park et al. 2010b). The loss of the E-cadherin is regarded as a key step in initiation of EMT (Thiery et al. 2009) and now it is clear that Snail, E47 and Zebs can directly bind to the promoter of E-cadherin, thereby repressing expression of E-cadherin (Peinado et al. 2007). There are several potential links between hyper-O-GlcNAcylation, repression of E-cadherin expression, and enhancement of invasion and metastasis. O-GlcNAcylation of E-cadherin in its cytoplasmic domain during ER stress blocks its cell surface transport, thereby inhibiting intercellular adhesion (Zhu et al. 2001), and thus potentially promoting migration/metastasis. Also, direct O-GlcNAcylation of Snail on Ser112 stabilizes it, increasing repression of E-cadherin expression, and increasing cancer cell invasion and metastasis (Park et al. 2010a). Finally, the stability of Snail can be indirectly enhanced by NF-κB activity (Wu et al. 2009), suggesting that cancer cell associated increased NF-κB activity due to hyper-O-GlcNAcylation (Ma et al. 2013) may contribute to Snail stability, and thus metastasis.
Potential links between hyper-O-GlcNAcylation and aneuploidy in cancer
Genome instability, which is characterized by genetic aberrations from single nucleotide changes in genes to structural chromosomal abnormalities, also known as chromosomal instability (CIN), is another hallmark of cancer (Hanahan and Weinberg 2011; Gordon et al. 2012; McGranahan et al. 2012). Both CIN and aneuploidy are implicated in tumorigenesis. Cell cycle specific dynamic patterns of O-GlcNAcylation are linked to proper mitotic progression and cytokinesis. Overexpression of OGT leads to a polyploid phenotype due to severe defects in mitotic progression and cytokinesis, whereas reducing O-GlcNAc by overexpression of OGA induces a mitotic exit phenotype (Slawson et al. 2005). A recent study has shown that over 100 proteins involved in spindle assembly and cytokinesis are O-GlcNAcylated (Wang et al. 2010). While quite speculative, it is possible that hyper-O-GlcNAcylation may contribute to defects in chromosomal segregation due to spindle defects, which may contribute to polyploidy in cancer.
Possible connections between hyper-O-GlcNAcylation and the inflammatory tumor microenvironment
Infiltrating immune cells are general features of tumor microenvironments. Lymphocytes and neutrophils are two main immune cells that are associated with the tumor microenvironment (Mueller and Fusenig 2004). Cancer cell secretion of cytokines such as TGF-β, IL-6 and TNFα are thought to play a role in promoting this inflammatory tumor microenvironment by activating/recruiting lymphocytes and neutrophils. The transforming growth factor (TGF)-β-activated kinase 1 (TAK1) was implicated in cell transformation, in particular in pancreatic cancer where TAK1 is required for Kras transformation linked to NF-κB activation and downstream cytokine production and release (Bang et al. 2013). TAB 1 binding to TAK1 is required for activation. TAB 1 is O-GlcNAc modified at serine 359, and this post-translational event is required for TAK1 activation and downstream NF-κB activity leading to IL-6 and TNFα production in response to IL-1 signaling (Pathak et al. 2012). We have also shown that hyper-O-GlcNAcylation of NF-κB signaling pathway components p65 and IKKs are associated with oncogenic up-regulation of NF-κB (Ma et al. 2013). Taken together, the results suggest that hyper-O-GlcNAcylation of TAB 1and/or NF-κB pathway components may play a role in promoting an inflamatory tumor microenvironment through production/release of inflammatory cytokines.
Hyper-O-GlcNAcylation and cancer stem cells
Most tumors, particularly solid tumors, are composed of heterogeneous cell populations, including a newly appreciated subclass of tumor cells, known as cancer stem cells (CSCs) (Alison et al. 2012; Hanahan and Weinberg 2011). CSCs display uniquely high rates of self-renewal and have superior potential to produce new tumors upon in vivo xenograft (Alison et al. 2012; Hanahan and Weinberg 2011). O-GlcNAc has been shown to modify several transcriptional factors such as c-Myc, Oct4, Sox2 (Jang et al. 2012; Myers et al. 2011; Chou et al. 1995b), which are implicated in the regulation of pluripotency and in reprogramming somatic cells into induced pluripotent stem cells (iPSCs) (Vierbuchen and Wernig 2012). Upon embryonic stem cell differentiation, O-GlcNAcylation is quickly reduced on Sox2 and Oct4 (Jang et al. 2012). While the roles for O-GlcNAcylation on Sox2 are not clear yet, O-GlcNAcylation on Oct4 at T228 is important to maintain embryonic stem cell self-renewal, reprogram somatic cells and induce several pluripotency-related genes (Jang et al. 2012). These results indicate that hyper-O-GlcNAcylation in cancer may contribute to CSC self-renewal and pluripotency.
Hyper-O-GlcNAcylation may contribute to metabolic reprogramming in cancer
Due to responsiveness of O-GlcNAcylation to glucose flux, O-GlcNAc was termed a “nutritional sensor” and may provide feedback signals that modulate metabolism in response to changing cellular nutrient status. Several studies now link hyper-O-GlcNAcylation to cancer-associated metabolic reprogramming (Fig. 3).

Hyper-O-GlcNAcylation promotes cancer cell metabolic reprogramming. In non-transformed cells, glucose mainly fluxes through the TCA cycle and oxidative phosphorylation produces energy. In cancer cells, the main energy source shifts to aerobic glycolysis, which is programed by oncogenes such as HIF1α, Kras, and c-Myc. This metabolic reprogramming is facilitated by hyper-O-GlcNAcylation in cancer. Cancer cell-specific O-GlcNAcylation of PFK1 at S529 inhibits its kinase activity in cancer cells. As a result, more glycolytic intermediates shunt into branch pathways such as the PPP and HBP, further driving elevated O-GlcNAcylation. Also, elevation of O-GlcNAcylation increases hexokinase activity and decreases pyruvate kinase activity, which is consistent with increasing glucose flux through glycolytic branch pathways. In addition, ChREBP and c-Myc are stabilized by O-GlcNAcylation. ChREBP is implicated in upregulating aerobic glycolysis, de novo lipogenesis and nucleotide biosynthesis. c-Myc is also involved in upregulation of aerobic glycolysis and glutamine metabolism. The thickness of arrows indicates relative rates of flux in pathways. Proteins in red are increased in the level of expression, stability, or activity by hyper-O-GlcNAcylation and those in green are decreased in expression or activity. “G” denotes O-GlcNAcylation. ChREBP carbohydrate responsive element-binding protein, FAS fatty acid synthase, HK hexokinase, HBP hexosamine biosynthetic pathway, G6P Glucose-6-phosphate, F6P fructose-6-phosphate, FBP fructose 1,6-bisphosphate, PEP phosphoenolpyruvate, GFAT glutamine: fructose-6-phosphate amidotransferase, GSH reduced glutathione, GSSG oxidized glutathione, PFK1 phosphofructokinase 1, PK pyruvate kinase, PPP pentose phosphate pathway (color figure online)
O-GlcNAc has been reported to modify a variety of glycolytic enzymes, including glucose-6-phosphate isomerase (GPI), phosphofructokinase 1 (PFK1), fructose-bisphosphate aldolase A (ALDOA), triosephosphate isomerase (TPI), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase 2 (PGK2), alpha-enolase (Eno1), pyruvate kinase isozymes M2 (PKM2), lactate dehydrogenase A chain (LDHA) (Nandi et al. 2006; Ohn et al. 2008; Teo et al. 2010). Recently, O-GlcNAc has been mapped at serine 529 on PFK1, which catalyzes the rate-limiting step of glycolysis to generate fructose-1, 6-bisphosphate (F1, 6BP) from fructose-6-phosphate (F6P), and the role for O-GlcNAc on PFK1 has been elucidated (Yi et al. 2012). In order to be fully active, PFK1 forms tetramers or even higher oligomers and serine 529 is vital for allosteric activation of PFK1 by F2, 6BP (Sola-Penna et al. 2010). O-GlcNAcylation of PFK1 at serine 529 inhibits its kinase activity in cancer cells, possibly resulting from the O-GlcNAc moiety blocking the binding of F2, 6BP to PFK1 and disrupting oligomerization of PFK1 (Yi et al. 2012), leading to increased levels of glycolytic intermediates. This impacts cancer cells in several ways: first, flux through the PPP is increased leading to increased supplies of nucleotides for cancer cell proliferation; second, increased PPP flux helps cancer cells cope with oxidative stress as PPP generated NADPH helps to keep anti-oxidant GSH in a reduced state (Yi et al. 2012); third, flux through the HBP is increased which could further elevate O-GlcNAc by increasing UDP-GlcNAc levels. Importantly, O-GlcNAcylation of PFK1 is only increased in transformed cells, but not in highly proliferating normal T cells or epithelial cells (Yi et al. 2012). These data suggest that PFK1 could be a cancer-specific target. Another glycolytic enzyme with established links to cancer is PKM2, which catalyzes conversion of phosphoenolpyruvate (PEP) to pyruvate. PKM2 is almost universally expressed in highly proliferating cells like cancer cells, while PKM1 is predominantly expressed in most adult differentiated tissues. While PKM1 is constitutively active, PKM2 has intrinsically lower enzymatic activity and is sensitive to inhibition by receptor kinase signaling (Kroemer and Pouyssegur 2008; Ward and Thompson 2012). Thus, it seems counterintuitive that PKM2 is employed in highly proliferating cells. However, it was suggested that the lower enzymatic activity of PKM2 limits conversion of glycolytic intermediates to pyruvate, thereby shunting more of these intermediates into anabolic biosynthetic pathways to support cell proliferation and growth (Ward and Thompson 2012; Kroemer and Pouyssegur 2008). Although the site and function of O-GlcNAcylation on PKM2 is not established, increasing O-GlcNAcylaiton in general causes a significant decrease in pyruvate kinase activity (Yi et al. 2012). Thus, cancer cell hyper-O-GlcNAcylation would likely decrease PKM2 activity, further contributing to directing glycolytic intermediates away from pyruvate and toward biosynthetic pathways. Additionally, elevating O-GlcNAcylation increases the enzymatic activity of hexokinase (Yi et al. 2012), which catalyzes the first step of glycolysis. In this case, cancer cell hyper-O-GlcNAcylation may increase hexokinase activity thus trapping more glucose in cancer cells and increasing shunt into pathways such as the PPP and HBP. These data suggest that cancer cell hyper-O-GlcNAcylation may regulate glycolytic enzymes in such a way as to contribute to cancer cell increased consumption and flux of glucose through biosynthetic branch pathways, thereby contributing to metabolic reprogramming in cancer. Thus, hyper-O-GlcNAcylation may not only be a consequence of altered cancer metabolism, but also participate in a cycle in which hyper-O-GlcNAcylation itself drives cancer metabolism phenotypes, possibly further acerbating hyper-O-GlcNAcylation.
In addition to modifying enzymes involved in glycolysis, O-GlcNAcylation may also regulate transcription factors to modulate metabolic reprogramming. Carbohydrate responsive element-binding protein (ChREBP), a basic helix–loop–helix leucine zipper transcription factor, is essential for the induction of the glycolytic enzyme L-pyruvate kinase (L-PK) and lipogenic genes [e.g., acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS)] in response to glucose. ChREBP plays a crucial role in regulating energy metabolism and its deregulation can contribute to metabolic diseases (Havula and Hietakangas 2012). Recently, it has been shown that ChREBP is required for efficient cancer cell proliferation and that suppression of ChREBP leads to decreased aerobic glycolysis, de novo lipogenesis and nucleotide biosynthesis, and to increased mitochondrial respiration, indicating activated ChREBP at least in part contributes to metabolic reprogramming in tumors (Tong et al. 2009). ChREBP has been reported to be O-GlcNAcylated (Sakiyama et al. 2010; Guinez et al. 2011). O-GlcNAcylation leads to the stabilization of ChREBP protein and increased transcription of ChREBP target genes L-PK, ACC and FAS. Conversely, lowering O-GlcNAcylation of ChREBP decreases levels of the lipogenic proteins ACC and FAS and prevents hepatic steatosis (Guinez et al. 2011). Although the O-GlcNAc sites on ChREBP was not mapped, cancer cell hyper-O-GlcNAcylation may stabilize/activate ChREBP, driving cancer cell increased aerobic glycolysis and lipogenesis, and representing a mechanism that could contribute to hyper-O-GlcNAcylation support of transformed phenotypes.
Another potential link between hyper-O-GlcNAcylation and cancer metabolism is c-Myc. Oncogenic c-Myc is a transcription factor that is not only involved in tumor initiation and maintenance, but also up-regulates glycolysis and promotes mitochondrial metabolism and mitochondrial biogenesis (Ward and Thompson 2012). c-Myc induces the expression of glycolytic enzymes and LDHA and increases the expression of glutaminase that converts glutamine to glutamate in mitochondria for anapleurotic resupply of TCA intermediates used in biosynthesis (DeBerardinis et al. 2008). c-Myc is subject to phosphorylation at Thr58 by GSK3β (Gregory et al. 2003). Phosphorylation at Thr58 causes the rapid degradation of c-Myc and reduces its target gene expression (Vervoorts et al. 2006). Thr58 is located in the transactivation domain and is a mutational hot spot in many lymphomas, leading to the stabilization of c-Myc (Chou et al. 1995b). c-Myc is also O-GlcNAcylated at Thr58 (Chou et al. 1995a). Thus, increased c-myc Thr58 O-GlcNAcylation could compete with phosphorylation and potentially stabilize c-Myc. Indeed, reducing O-GlcNAc has been shown to cause the degradation of c-Myc protein but not the mRNA in prostate cancer cells by an OGT inhibitor (Itkonen et al. 2013). These results suggest that hyper-O-GlcNAcylation may contribute to oncogenicity and cancer metabolic reprograming through stabilizing oncogenic c-Myc.
Therapeutic potential of targeting cancer hyper-O-GlcNAcylation
Key aspects of treating human cancer will involve reliable biomarkers for early diagnosis, prediction of prognosis, and detection of recurrence. Seminal studies have demonstrated that OGT and OGA may serve as biomarkers for early detection and prognosis. OGT mRNA is detected in 51.7 % of 176 urine samples obtained from bladder cancer patients, but not detected in 143 healthy individuals (Rozanski et al. 2012). Furthermore, the mRNA expression of OGT is correlated with the differentiation of bladder tumor, with the poorly differentiated (grade III) most aggressive form manifesting the highest OGT mRNA levels (Rozanski et al. 2012). Although OGA mRNA is found in urine from bladder cancer patients and healthy individuals with a comparable rate, mRNA levels of OGA are inversely correlated with the differentiation state of bladder tumor (Rozanski et al. 2012). These results are consistent with our findings in prostate and pancreatic cancer cells. In addition, low levels of OGA correlate with tumor recurrence in hepatocellular carcinoma (HCC) patients receiving liver transplantation (Zhu et al. 2012). Moreover, O-GlcNAc levels in blood and spleen can monitor the changes in tumor burden of chronic lymphocytic leukemia (Tomic et al. 2013). In the 1950s, glutamine analogs such as 6-diazo-5-oxo-L-norleucine (DON) and azaserine, which are now known to inhibit the HBP rate limiting enzyme GFAT (Badet et al. 1987), were found to inhibit cancer cell growth (Moore and Lepage 1957; Tarnowski and Stock 1957). A remarkable number of studies examined the efficacy of glutamine analogs in blocking transformed cell growth (Kisner et al. 1980; Livingston et al. 1970). In a variety of human xenograft tumors transplanted in athymic mice, including colon, mammary, and lung, DON induces remarkable tumor regressions (Ovejera et al. 1979). Given the success of these glutamine analogs in vitro, DON and azaserine were tested in a variety of human cancer clinical trials (Earhart et al. 1990; Lynch et al. 1982) and found to be effective in reducing tumor burden, but also had severe intolerable side-effects of vomiting and nausea, which limited enthusiasm for their use as chemotherapeutic agents. Targeting the HBP would be expected to influence any glycosylation processes dependent on HBP end products UDP-GlcNAc and UDP-GalNAc (perhaps accounting for the toxicity observed). Nevertheless, it is tempting to speculate that anti-cancer effects of inhibition of the HBP may have involved the consequent reduction of hyper-O-GlcNAcylation that would have occurred. A key aspect of targeting OGT as an anti-cancer approach is its apparent cancer cell selectivity of inhibiting growth. As was discussed above, genetically targeting hyper-O-GlcNAcylation by knocking down OGT inhibits many hallmarks of cancer, but does not affect the cell proliferation of immortalized non-transformed cells derived from breast, prostate and pancreas (Caldwell et al. 2010; Lynch et al. 2012; Ma et al. 2013), which suggests that OGT may be cancer specific target. Ongoing efforts are attempting to identify effective small molecule specific inhibitors of OGT. One way to target OGT is to develop cell-permeable small-molecule inhibitors. The first reported OGT inhibitors were discovered by high-throughput screens based on ligand displacement assay (Gross et al. 2005). These inhibitors are benzoxazolinone derivatives which inhibit intracellular O-GlcNAcylation in cell culture and inhibit breast cancer cell growth in vitro (Caldwell et al. 2010). However, the relatively low efficacy of OGT inhibition requires use at relatively high levels (500 μm in cell culture) and they display poor water solubility, impeding use for animal studies. O-GlcNAc analog competitive inhibitors of OGT, 5-thioglucosamine (5SGlcNAc) and its per-O-acetylated analog Ac-5SGlcNAc were developed (Gloster et al. 2011). They can be converted into UDP-5SGlcNAc via the GlcNAc salvage pathway, thereby competing with UDP-GlcNAc (Gloster et al. 2011). We have shown that Ac-5SGlcNAc is able to reduce cellular O-GlcNAc and inhibit pancreatic cancer cell growth in vitro (Ma et al. 2013). However, future work will need to involve screening and design of more bioavailable and efficacious OGT inhibitors.
Conclusion and therapeutic perspective
Hallmarks of cancer provide a framework to review potential connections between hyper-O-GlcNAcylation and the complexities of transformed cell states. As discussed above, elevation of O-GlcNAc in cancer on specific proteins may be linked to many if not all hallmarks of cancer. Similar to “oncogene addiction” in cancers, cancer cells appear to display an “addiction” to hyper-O-GlcNAcylation, as targeting OGT to reduce hyper-O-GlcNAcylation inhibits cancer cell growth while non-cancer cell growth is not altered. Thus, abnormal hyper-O-GlcNAcylation may be an attractive novel cancer specific therapeutic target. With the development of more bioavailable and efficacious OGT inhibitors, it is expected that suppression of hyper-O-GlcNAcylation by targeting OGT may serve as a novel therapeutic intervention for a variety of cancers.
Abbreviations
- HBP:
-
Hexosamine biosynthetic pathway
- GlcNAc:
-
N-acetylglucosamine
- PDAC:
-
Pancreatic ductal adenocarcinoma
- OGA:
-
O-GlcNAcase
- HPDE:
-
Human pancreatic duct epithelial cell
- PPP:
-
Pentose phosphate pathway
- HSR:
-
Heat shock response
- EMT:
-
Epithelial to mesenchymal transition
References
Alison MR, Lin WR, Lim SM, Nicholson LJ (2012) Cancer stem cells: in the line of fire. Cancer Treat Rev 38(6):589–598. doi:10.1016/j.ctrv.2012.03.003
Andres-Bergos J, Tardio L, Larranaga-Vera A, Gomez R, Herrero-Beaumont G, Largo R (2012) The increase in O-linked N-acetylglucosamine protein modification stimulates chondrogenic differentiation both in vitro and in vivo. J Biol Chem 287(40):33615–33628. doi:10.1074/jbc.M112.354241
Badet B, Vermoote P, Haumont PY, Lederer F, LeGoffic F (1987) Glucosamine synthetase from Escherichia coli: purification, properties, and glutamine-utilizing site location. Biochemistry 26(7):1940–1948
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19(5):329–337. doi:10.1016/j.semcancer.2009.05.003
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319(5865):916–919. doi:10.1126/science.1141448
Bang D, Wilson W, Ryan M, Yeh JJ, Baldwin AS (2013) GSK-3alpha promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-κB. Cancer discov 3(6):690–703. doi:10.1158/2159-8290.cd-12-0541
Benz CC, Yau C (2008) Ageing, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer 8(11):875–879. doi:10.1038/nrc2522
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2(10):737–744. doi:10.1038/35036374
Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, Reginato MJ (2010) Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 29(19):2831–2842. doi:10.1038/onc.2010.41
Chou TY, Dang CV, Hart GW (1995a) Glycosylation of the c-Myc transactivation domain. Proc Natl Acad Sci U S A 92(10):4417–4421
Chou TY, Hart GW, Dang CV (1995b) c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem 270(32):18961–18965
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130(6):1005–1018. doi:10.1016/j.cell.2007.07.020
Dai C, Dai S, Cao J (2012) Proteotoxic stress of cancer: implication of the heat-shock response in oncogenesis. J Cell Physiol 227(8):2982–2987. doi:10.1002/jcp.24017
DeBerardinis RJ (2008) Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med 10(11):767–777. doi:10.1097/GIM.0b013e31818b0d9b
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20. doi:10.1016/j.cmet.2007.10.002
Dudeja V, Chugh RK, Sangwan V, Skube SJ, Mujumdar NR, Antonoff MB, Dawra RK, Vickers SM, Saluja AK (2011) Prosurvival role of heat shock factor 1 in the pathogenesis of pancreatobiliary tumors. Am J Phys Gastrointest Liver Physiol 300(6):G948–G955. doi:10.1152/ajpgi.00346.2010
Earhart RH, Amato DJ, Chang AY, Borden EC, Shiraki M, Dowd ME, Comis RL, Davis TE, Smith TJ (1990) Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Invest New Drugs 8(1):113–119
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3(5):362–374. doi:10.1038/nrc1075
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458(7239):762–765. doi:10.1038/nature07823
Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ (2011) Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7(3):174–181. doi:10.1038/nchembio.520
Gong J, Jing L (2011) Glutamine induces heat shock protein 70 expression via O-GlcNAc modification and subsequent increased expression and transcriptional activity of heat shock factor-1. Minerva Anestesiol 77(5):488–495
Gordon DJ, Resio B, Pellman D (2012) Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13(3):189–203. doi:10.1038/nrg3123
Gregory MA, Qi Y, Hann SR (2003) Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 278(51):51606–51612. doi:10.1074/jbc.M310722200
Gross BJ, Kraybill BC, Walker S (2005) Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc 127(42):14588–14589. doi:10.1021/ja0555217
Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C, Yang J, Han F, Lu X, Yu W (2010) GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res 70(15):6344–6351. doi:10.1158/0008-5472.can-09-1887
Guinez C, Lemoine J, Michalski JC, Lefebvre T (2004) 70-kDa-heat shock protein presents an adjustable lectinic activity towards O-linked N-acetylglucosamine. Biochem Biophys Res Commun 319(1):21–26. doi:10.1016/j.bbrc.2004.04.144
Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, Moldes M, Burnol AF, Yang X, Lefebvre T, Girard J, Postic C (2011) O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60(5):1399–1413. doi:10.2337/db10-0452
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695. doi:10.1016/j.cell.2006.11.001
Hallor KH, Sciot R, Staaf J, Heidenblad M, Rydholm A, Bauer HC, Astrom K, Domanski HA, Meis JM, Kindblom LG, Panagopoulos I, Mandahl N, Mertens F (2009) Two genetic pathways, t(1;10) and amplification of 3p11-12, in myxoinflammatory fibroblastic sarcoma, haemosiderotic fibrolipomatous tumour, and morphologically similar lesions. J Pathol 217(5):716–727. doi:10.1002/path.2513
Haltiwanger RS, Blomberg MA, Hart GW (1992) Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase. J Biol Chem 267(13):9005–9013
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86(3):353–364
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Hart GW (1997) Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu Rev Biochem 66:315–335. doi:10.1146/annurev.biochem.66.1.315
Hart GW, Housley MP, Slawson C (2007) Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446(7139):1017–1022. doi:10.1038/nature05815
Havula E, Hietakangas V (2012) Glucose sensing by ChREBP/MondoA-Mlx transcription factors. Semin Cell Dev Biol 23(6):640–647. doi:10.1016/j.semcdb.2012.02.007
Ho SR, Wang K, Whisenhunt TR, Huang P, Zhu X, Kudlow JE, Paterson AJ (2010) O-GlcNAcylation enhances FOXO4 transcriptional regulation in response to stress. FEBS Lett 584(1):49–54. doi:10.1016/j.febslet.2009.11.059
Itkonen HM, Minner S, Guldvik IJ, Sandmann MJ, Tsourlakis MC, Berge V, Svindland A, Schlomm T, Mills IG (2013) O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer. Cancer Res. doi:10.1158/0008-5472.can-13-0549
Jang H, Kim TW, Yoon S, Choi SY, Kang TW, Kim SY, Kwon YW, Cho EJ, Youn HD (2012) O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 11(1):62–74. doi:10.1016/j.stem.2012.03.001
Kawauchi K, Araki K, Tobiume K, Tanaka N (2008) p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol 10(5):611–618. doi:10.1038/ncb1724
Kawauchi K, Araki K, Tobiume K, Tanaka N (2009) Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A 106(9):3431–3436. doi:10.1073/pnas.0813210106
Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE (2010) O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. J Biol Chem 285(50):39096–39107. doi:10.1074/jbc.M110.131102
Kisner DL, Catane R, Muggia FM (1980) The rediscovery of DON (6-diazo-5-oxo-L-norleucine). Recent Results Cancer Res 74:258–263
Kreppel LK, Hart GW (1999) Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J Biol Chem 274(45):32015–32022
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13(6):472–482. doi:10.1016/j.ccr.2008.05.005
Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S (2011) Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature 469(7331):564–567. doi:10.1038/nature09638
Liu J, Marchase RB, Chatham JC (2007) Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J Mol Cell Cardiol 42(1):177–185. doi:10.1016/j.yjmcc.2006.09.015
Livingston RB, Venditti JM, Cooney DA, Carter SK (1970) Glutamine antagonists in chemotherapy. Adv Pharmacol Chemother 8:57–120
Luo J, Solimini NL, Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136(5):823–837. doi:10.1016/j.cell.2009.02.024
Lynch G, Kemeny N, Casper E (1982) Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol 5(5):541–543
Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ (2012) Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 287(14):11070–11081. doi:10.1074/jbc.M111.302547
Ma Z, Vocadlo DJ, Vosseller K (2013) Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J Biol Chem 288(21):15121–15130. doi:10.1074/jbc.M113.470047
Manzari B, Kudlow JE, Fardin P, Merello E, Ottaviano C, Puppo M, Eva A, Varesio L (2007) Induction of macrophage glutamine: fructose-6-phosphate amidotransferase expression by hypoxia and by picolinic acid. Intern J Immunopathol Pharmacol 20(1):47–58
Mariappa D, Sauert K, Marino K, Turnock D, Webster R, van Aalten DM, Ferguson MA, Muller HA (2011) Protein O-GlcNAcylation is required for fibroblast growth factor signaling in Drosophila. Sci Signal 4 (204): ra89. doi:10.1126/scisignal.2002335
Marotta NP, Cherwien CA, Abeywardana T, Pratt MR (2012) O-GlcNAc modification prevents peptide-dependent acceleration of α-synuclein aggregation. Chembiochem. doi:10.1002/cbic.201200478
Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192(1):1–15. doi:10.1002/jcp.10119
McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C (2012) Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 13(6):528–538. doi:10.1038/embor.2012.61
Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X, Cong Q, Yu W (2011) O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta 1812(4):514–519. doi:10.1016/j.bbadis.2011.01.009
Moore EC, Lepage GA (1957) In vivo sensitivity of normal and neoplastic mouse tissues to azaserine. Cancer Res 17(8):804–808
Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E (2007) Energy metabolism in tumor cells. FEBS J 274(6):1393–1418. doi:10.1111/j.1742-4658.2007.05686.x
Mueller MM, Fusenig NE (2004) Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 4(11):839–849. doi:10.1038/nrc1477
Myatt SS, Lam EW (2007) The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 7(11):847–859. doi:10.1038/nrc2223
Myers SA, Panning B, Burlingame AL (2011) Polycomb repressive complex 2 is necessary for the normal site-specific O-GlcNAc distribution in mouse embryonic stem cells. Proc Natl Acad Sci U S A 108(23):9490–9495. doi:10.1073/pnas.1019289108
Nandi A, Sprung R, Barma DK, Zhao Y, Kim SC, Falck JR (2006) Global identification of O-GlcNAc-modified proteins. Anal Chem 78(2):452–458. doi:10.1021/ac051207j
Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18(1):64–76. doi:10.1158/1078-0432.ccr-11-1000
Ngoh GA, Watson LJ, Facundo HT, Jones SP (2011) Augmented O-GlcNAc signaling attenuates oxidative stress and calcium overload in cardiomyocytes. Amino Acids 40(3):895–911. doi:10.1007/s00726-010-0728-7
O’Donnell N, Zachara NE, Hart GW, Marth JD (2004) Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol 24(4):1680–1690
Ohn T, Kedersha N, Hickman T, Tisdale S, Anderson P (2008) A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat Cell Biol 10(10):1224–1231. doi:10.1038/ncb1783
Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275(29):21797–21800. doi:10.1074/jbc.C000023200
Ovejera AA, Houchens DP, Catane R, Sheridan MA, Muggia FM (1979) Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Res 39(8):3220–3224
Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, Ota I, Shimada K, Konishi N, Nam HW, Hong SW, Yang WH, Roth J, Yook JI, Cho JW (2010a) Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. doi:10.1038/emboj.2010.254
Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, Ota I, Shimada K, Konishi N, Nam HW, Hong SW, Yang WH, Roth J, Yook JI, Cho JW (2010b) Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J 29(22):3787–3796. doi:10.1038/emboj.2010.254
Pathak S, Borodkin VS, Albarbarawi O, Campbell DG, Ibrahim A, van Aalten DM (2012) O-GlcNAcylation of TAB 1 modulates TAK1-mediated cytokine release. EMBO J 31(6):1394–1404. doi:10.1038/emboj.2012.8
Peinado H, Olmeda D, Cano A (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7(6):415–428. doi:10.1038/nrc2131
Perkins ND (2012) The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer 12(2):121–132. doi:10.1038/nrc3204
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radical Biol Med 49(11):1603–1616. doi:10.1016/j.freeradbiomed.2010.09.006
Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM (2007) Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9(2):166–180
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM (2004) Oncomine: a cancer microarray database and integrated data-mining platform. Neoplasia 6(1):1–6
Roos MD, Han IO, Paterson AJ, Kudlow JE (1996) Role of glucosamine synthesis in the stimulation of TGF-alpha gene transcription by glucose and EGF. Am J Physiol 270(3 Pt 1):C803–C811
Rozanski W, Krzeslak A, Forma E, Brys M, Blewniewski M, Wozniak P, Lipinski M (2012) Prediction of bladder cancer based on urinary content of MGEA5 and OGT mRNA level. Clin Lab 58(5–6):579–583
Sakiyama H, Fujiwara N, Noguchi T, Eguchi H, Yoshihara D, Uyeda K, Suzuki K (2010) The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem Biophys Res Commun 402(4):784–789. doi:10.1016/j.bbrc.2010.10.113
Shi Y, Tomic J, Wen F, Shaha S, Bahlo A, Harrison R, Dennis JW, Williams R, Gross BJ, Walker S, Zuccolo J, Deans JP, Hart GW, Spaner DE (2010) Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia 24(9):1588–1598. doi:10.1038/leu.2010.152
Singleton KD, Wischmeyer PE (2008) Glutamine induces heat shock protein expression via O-glycosylation and phosphorylation of HSF-1 and Sp1. JPEN J Parenter Enteral Nutr 32(4):371–376. doi:10.1177/0148607108320661
Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW (2005) Perturbations in O-linked beta-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem 280(38):32944–32956. doi:10.1074/jbc.M503396200
Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM, Zancan P (2010) Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life 62(11):791–796. doi:10.1002/iub.393
Tarnowski GS, Stock CC (1957) Effects of combinations of azaserine and of 6-diazo-5-oxo-L-norleucine with purine analogs and other antimetabolites on the growth of two mouse mammary carcinomas. Cancer Res 17(10):1033–1039
Teo CF, Ingale S, Wolfert MA, Elsayed GA, Not LG, Chatham JC, Wells L, Boons GJ (2010) Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat Chem Biol 6(5):338–343. doi:10.1038/nchembio.338
Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7(2):131–142. doi:10.1038/nrm1835
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139(5):871–890. doi:10.1016/j.cell.2009.11.007
Tomic J, McCaw L, Li Y, Hough MR, Ben-David Y, Moffat J, Spaner DE (2013) Resveratrol has anti-leukemic activity associated with decreased O-Glcnacylated proteins. Exp Hematol. doi:10.1016/j.exphem.2013.04.004
Tong X, Zhao F, Mancuso A, Gruber JJ, Thompson CB (2009) The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc Natl Acad Sci U S A 106(51):21660–21665. doi:10.1073/pnas.0911316106
Torres CR, Hart GW (1984) Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 259(5):3308–3317
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147(2):275–292. doi:10.1016/j.cell.2011.09.024
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033. doi:10.1126/science.1160809
Vervoorts J, Luscher-Firzlaff J, Luscher B (2006) The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem 281(46):34725–34729. doi:10.1074/jbc.R600017200
Vierbuchen T, Wernig M (2012) Molecular roadblocks for cellular reprogramming. Mol Cell 47(6):827–838. doi:10.1016/j.molcel.2012.09.008
Walgren JL, Vincent TS, Schey KL, Buse MG (2003) High glucose and insulin promote O-GlcNAc modification of proteins, including alpha-tubulin. Am J Physiol Endocrinol Metabol 284(2):E424–E434. doi:10.1152/ajpendo.00382.2002
Wang Z, Udeshi ND, Slawson C, Compton PD, Sakabe K, Cheung WD, Shabanowitz J, Hunt DF, Hart GW (2010) Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci Signal 3 (104):ra2. doi:10.1126/scisignal.2000526
Warburg O (1956a) On respiratory impairment in cancer cells. Science 124(3215):269–270
Warburg O (1956b) On the origin of cancer cells. Science 123(3191):309–314
Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J General Physiol 8(6):519–530
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21(3):297–308. doi:10.1016/j.ccr.2012.02.014
Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17(11):1359–1370. doi:10.1038/nm.2537
Wells L, Vosseller K, Hart GW (2001) Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science 291(5512):2376–2378
Wells L, Vosseller K, Hart GW (2003) A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci 60(2):222–228
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105(48):18782–18787. doi:10.1073/pnas.0810199105
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP (2009) Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15(5):416–428. doi:10.1016/j.ccr.2009.03.016
Xia Y, Rocchi P, Iovanna JL, Peng L (2012) Targeting heat shock response pathways to treat pancreatic cancer. Drug Discov Today 17(1–2):35–43. doi:10.1016/j.drudis.2011.09.016
Yehezkel G, Cohen L, Kliger A, Manor E, Khalaila I (2012) O-GlcNAcylation in primary and metastatic colorectal cancer clones and effect of O-GlcNAcase silencing on cell phenotype and transcriptome. J Biol Chem. doi:10.1074/jbc.M112.345546
Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC (2012) Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337(6097):975–980. doi:10.1126/science.1222278
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149(3):656–670. doi:10.1016/j.cell.2012.01.058
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y (2007) Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178(1):93–105. doi:10.1083/jcb.200703099
Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ (2012) Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol 8(4):393–399. doi:10.1038/nchembio.797
Zachara NE, Hart GW (2004) O-GlcNAc a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim Biophy Acta 1673(1–2):13–28
Zachara NE, O’Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW (2004) Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem 279(29):30133–30142. doi:10.1074/jbc.M403773200
Zhu W, Leber B, Andrews DW (2001) Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J 20(21):5999–6007. doi:10.1093/emboj/20.21.5999
Zhu Q, Zhou L, Yang Z, Lai M, Xie H, Wu L, Xing C, Zhang F, Zheng S (2012) O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol 29(2):985–993. doi:10.1007/s12032-011-9912-1
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ma, Z., Vosseller, K. O-GlcNAc in cancer biology. Amino Acids 45, 719–733 (2013). https://doi.org/10.1007/s00726-013-1543-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-013-1543-8