Histone Displacement during Nucleotide Excision Repair

Abstract
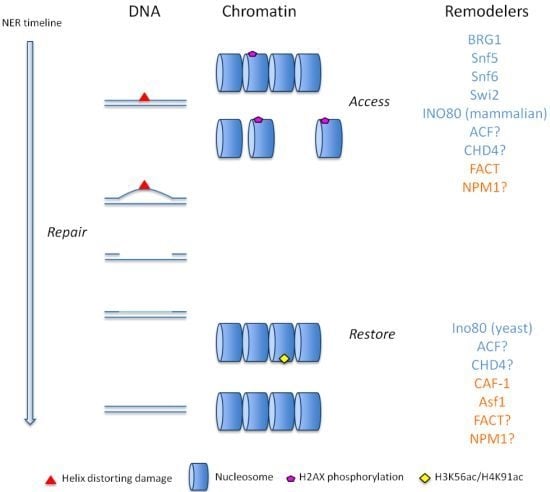
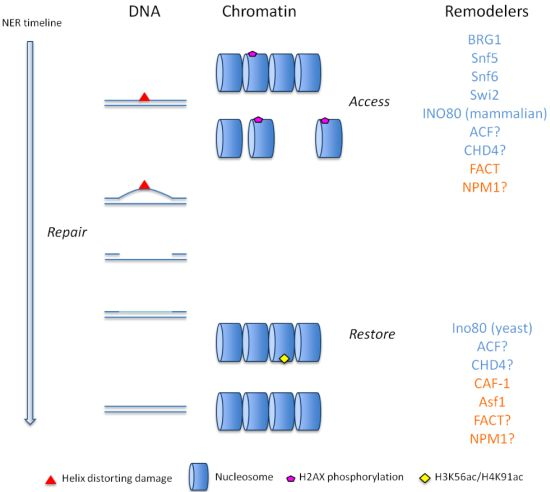
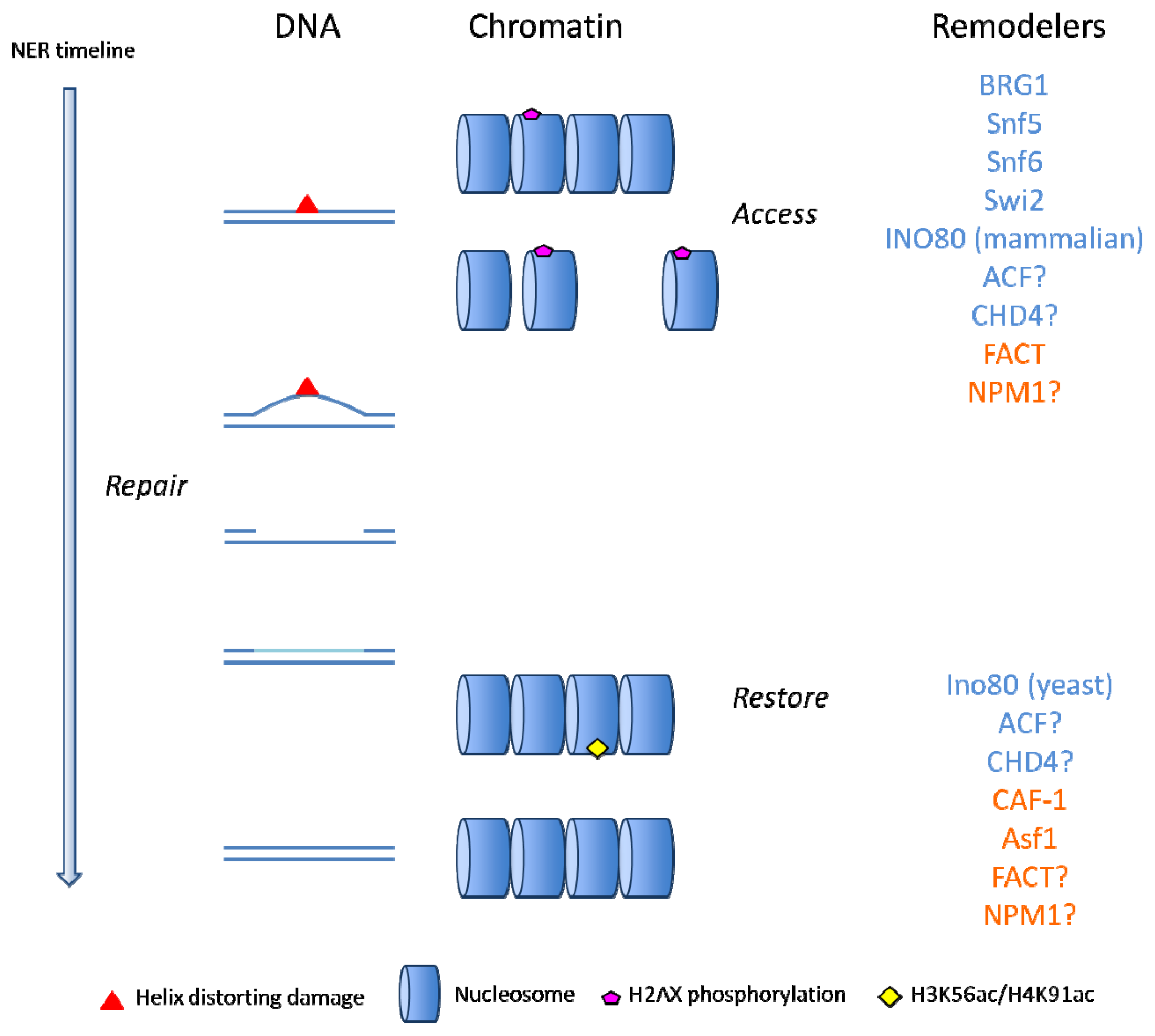
:
1. Chromatin
2. Nucleotide Excision Repair
3. ATP-Dependent Remodelers
4. Histone Chaperones
5. Conclusions
Acknowledgements
References
- Smerdon, M.J. DNA repair and the role of chromatin structure. Curr. Opin. Cell Biol 1991, 3, 422–428. [Google Scholar]
- Adkins, M.W.; Howar, S.R.; Tyler, J.K. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol. Cell 2004, 14, 657–666. [Google Scholar]
- Hwang, B.J.; Ford, J.M.; Hanawalt, P.C.; Chu, G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 1999, 96, 424–428. [Google Scholar]
- Zhao, Q.; Wang, Q.E.; Ray, A.; Wani, G.; Han, C.; Milum, K.; Wani, A.A. Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. J. Biol. Chem 2009, 284, 30424–30432. [Google Scholar]
- Dinant, C.; Houtsmuller, A.B.; Vermeulen, W. Chromatin structure and DNA damage repair. Epigenetics Chromatin 2008, 1, 9. [Google Scholar]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol 2011, 13, 1161–1169. [Google Scholar]
- Hopfner, K.P.; Gerhold, C.B.; Lakomek, K.; Wollmann, P. Swi2/Snf2 remodelers: Hybrid views on hybrid molecular machines. Curr. Opin. Struct. Biol 2012, 22, 225–233. [Google Scholar]
- Cai, Y.; Jin, J.; Gottschalk, A.J.; Yao, T.; Conaway, J.W.; Conaway, R.C. Purification and assay of the human INO80 and SRCAP chromatin remodeling complexes. Methods 2006, 40, 312–317. [Google Scholar]
- Ransom, M.; Dennehey, B.K.; Tyler, J.K. Chaperoning histones during DNA replication and repair. Cell 2010, 140, 183–195. [Google Scholar]
- Eitoku, M.; Sato, L.; Senda, T.; Horikoshi, M. Histone chaperones: 30 years from isolation to elucidation of the mechanisms of nucleosome assembly and disassembly. Cell. Mol. Life Sci 2008, 65, 414–444. [Google Scholar]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar]
- Formosa, T. The role of FACT in making and breaking nucleosomes. Biochim. Biophys. Acta 2012, 1819, 247–255. [Google Scholar]
- Takahata, S.; Yu, Y.; Stillman, D.J. FACT and Asf1 regulate nucleosome dynamics and coactivator binding at the HO promoter. Mol. Cell 2009, 34, 405–415. [Google Scholar]
- Nouspikel, T. DNA repair in mammalian cells: Nucleotide excision repair: variations on versatility. Cell. Mol. Life Sci 2009, 66, 994–1009. [Google Scholar]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar]
- Aboussekhra, A.; Biggerstaff, M.; Shivji, M.K.; Vilpo, J.A.; Moncollin, V.; Podust, V.N.; Protic, M.; Hubscher, U.; Egly, J.M.; Wood, R.D. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995, 80, 859–868. [Google Scholar]
- Mu, D.; Park, C.H.; Matsunaga, T.; Hsu, D.S.; Reardon, J.T.; Sancar, A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem 1995, 270, 2415–2418. [Google Scholar]
- Hara, R.; Mo, J.; Sancar, A. DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol 2000, 20, 9173–9181. [Google Scholar]
- Hara, R.; Sancar, A. The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol. Cell. Biol 2002, 22, 6779–6787. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Bekker-Jensen, S.; Mailand, N. The ubiquitin- and SUMO-dependent signaling response to DNA double-strand breaks. FEBS Lett 2011, 585, 2914–2919. [Google Scholar]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar]
- Marteijn, J.A.; Bekker-Jensen, S.; Mailand, N.; Lans, H.; Schwertman, P.; Gourdin, A.M.; Dantuma, N.P.; Lukas, J.; Vermeulen, W. Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol 2009, 186, 835–847. [Google Scholar]
- Hanasoge, S.; Ljungman, M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis 2007, 28, 2298–2304. [Google Scholar]
- Zhang, L.; Zhang, Q.; Jones, K.; Patel, M.; Gong, F. The chromatin remodeling factor BRG1 stimulates nucleotide excision repair by facilitating recruitment of XPC to sites of DNA damage. Cell Cycle 2009, 8, 3953–3959. [Google Scholar]
- Gong, F.; Fahy, D.; Smerdon, M.J. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat. Struct. Mol. Biol 2006, 13, 902–907. [Google Scholar]
- Yu, Y.; Teng, Y.; Liu, H.; Reed, S.H.; Waters, R. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 2005, 102, 8650–8655. [Google Scholar]
- Jiang, Y.; Wang, X.; Bao, S.; Guo, R.; Johnson, D.G.; Shen, X.; Li, L. INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 17274–17279. [Google Scholar]
- Sarkar, S.; Kiely, R.; McHugh, P.J. The Ino80 chromatin-remodeling complex restores chromatin structure during UV DNA damage repair. J. Cell Biol 2010, 191, 1061–1068. [Google Scholar]
- Luijsterburg, M.S.; Dinant, C.; Lans, H.; Stap, J.; Wiernasz, E.; Lagerwerf, S.; Warmerdam, D.O.; Lindh, M.; Brink, M.C.; Dobrucki, J.W.; et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol 2009, 185, 577–586. [Google Scholar]
- Sanchez-Molina, S.; Mortusewicz, O.; Bieber, B.; Auer, S.; Eckey, M.; Leonhardt, H.; Friedl, A.A.; Becker, P.B. Role for hACF1 in the G2/M damage checkpoint. Nucleic Acids Res 2011, 39, 8445–8456. [Google Scholar]
- Lans, H.; Marteijn, J.A.; Schumacher, B.; Hoeijmakers, J.H.; Jansen, G.; Vermeulen, W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet 2010, 6, e1000941. [Google Scholar]
- Luijsterburg, M.S.; Acs, K.; Ackermann, L.; Wiegant, W.W.; Bekker-Jensen, S.; Larsen, D.H.; Khanna, K.K.; van Attikum, H.; Mailand, N.; Dantuma, N.P. A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure. EMBO J 2012, 31, 2511–2527. [Google Scholar]
- Polo, S.E.; Roche, D.; Almouzni, G. New histone incorporation marks sites of UV repair in human cells. Cell 2006, 127, 481–493. [Google Scholar]
- Mello, J.A.; Sillje, H.H.; Roche, D.M.; Kirschner, D.B.; Nigg, E.A.; Almouzni, G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep 2002, 3, 329–334. [Google Scholar]
- Miller, A.; Yang, B.; Foster, T.; Kirchmaier, A.L. Proliferating cell nuclear antigen and ASF1 modulate silent chromatin in Saccharomyces cerevisiae via lysine 56 on histone H3. Genetics 2008, 179, 793–809. [Google Scholar]
- Keller, D.M.; Lu, H. p53 serine 392 phosphorylation increases after UV through induction of the assembly of the CK2.hSPT16.SSRP1 complex. J. Biol. Chem 2002, 277, 50206–50213. [Google Scholar]
- Wu, M.H.; Chang, J.H.; Yung, B.Y. Resistance to UV-induced cell-killing in nucleophosmin/B23 over-expressed NIH 3T3 fibroblasts: enhancement of DNA repair and up-regulation of PCNA in association with nucleophosmin/B23 over-expression. Carcinogenesis 2002, 23, 93–100. [Google Scholar]
- Lans, H.; Marteijn, J.A.; Vermeulen, W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 2012, 5, 4. [Google Scholar]
- Luijsterburg, M.S.; Lindh, M.; Acs, K.; Vrouwe, M.G.; Pines, A.; van Attikum, H.; Mullenders, L.H.; Dantuma, N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol 2012, 197, 267–281. [Google Scholar]
- Fitch, M.E.; Nakajima, S.; Yasui, A.; Ford, J.M. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem 2003, 278, 46906–46910. [Google Scholar]
- Fei, J.; Kaczmarek, N.; Luch, A.; Glas, A.; Carell, T.; Naegeli, H. Regulation of nucleotide excision repair by UV-DDB: prioritization of damage recognition to internucleosomal DNA. PLoS Biol 2011, 9, e1001183. [Google Scholar]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 2010, 29, 3130–3139. [Google Scholar]
- Medina, P.P.; Sanchez-Cespedes, M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics 2008, 3, 64–68. [Google Scholar]
- Morrison, A.J.; Highland, J.; Krogan, N.J.; Arbel-Eden, A.; Greenblatt, J.F.; Haber, J.E.; Shen, X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004, 119, 767–775. [Google Scholar]
- Kashiwaba, S.; Kitahashi, K.; Watanabe, T.; Onoda, F.; Ohtsu, M.; Murakami, Y. The mammalian INO80 complex is recruited to DNA damage sites in an ARP8 dependent manner. Biochem. Biophys. Res. Commun 2010, 402, 619–625. [Google Scholar]
- van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar]
- Kitayama, K.; Kamo, M.; Oma, Y.; Matsuda, R.; Uchida, T.; Ikura, T.; Tashiro, S.; Ohyama, T.; Winsor, B.; Harata, M. The human actin-related protein hArp5: nucleo-cytoplasmic shuttling and involvement in DNA repair. Exp. Cell Res 2009, 315, 206–217. [Google Scholar]
- Conaway, R.C.; Conaway, J.W. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem. Sci 2009, 34, 71–77. [Google Scholar]
- Ura, K.; Araki, M.; Saeki, H.; Masutani, C.; Ito, T.; Iwai, S.; Mizukoshi, T.; Kaneda, Y.; Hanaoka, F. ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J 2001, 20, 2004–2014. [Google Scholar]
- Fyodorov, D.V.; Blower, M.D.; Karpen, G.H.; Kadonaga, J.T. Acf1 confers unique activities to ACF/CHRAC and promotes the formation rather than disruption of chromatin in vivo. Genes Dev 2004, 18, 170–183. [Google Scholar]
- Ito, T.; Levenstein, M.E.; Fyodorov, D.V.; Kutach, A.K.; Kobayashi, R.; Kadonaga, J.T. ACF consists of two subunits, Acf1 and ISWI, that function cooperatively in the ATP-dependent catalysis of chromatin assembly. Genes Dev 1999, 13, 1529–1539. [Google Scholar]
- Ito, T.; Bulger, M.; Pazin, M.J.; Kobayashi, R.; Kadonaga, J.T. ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 1997, 90, 145–155. [Google Scholar]
- Lan, L.; Ui, A.; Nakajima, S.; Hatakeyama, K.; Hoshi, M.; Watanabe, R.; Janicki, S.M.; Ogiwara, H.; Kohno, T.; Kanno, S.; et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol. Cell 2010, 40, 976–987. [Google Scholar]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Rendtlew Danielsen, J.M.; Menard, P.; Sand, J.C.; Stucki, M.; et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol 2010, 190, 731–740. [Google Scholar] [Green Version]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol 2010, 190, 741–749. [Google Scholar]
- Tyler, J.K.; Adams, C.R.; Chen, S.R.; Kobayashi, R.; Kamakaka, R.T.; Kadonaga, J.T. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 1999, 402, 555–560. [Google Scholar]
- Groth, A.; Ray-Gallet, D.; Quivy, J.P.; Lukas, J.; Bartek, J.; Almouzni, G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol. Cell 2005, 17, 301–311. [Google Scholar]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [Google Scholar]
- Green, C.M.; Almouzni, G. Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J 2003, 22, 5163–5174. [Google Scholar]
- Moggs, J.G.; Grandi, P.; Quivy, J.P.; Jonsson, Z.O.; Hubscher, U.; Becker, P.B.; Almouzni, G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol 2000, 20, 1206–1218. [Google Scholar]
- Sogo, J.M.; Stahl, H.; Koller, T.; Knippers, R. Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J. Mol. Biol 1986, 189, 189–204. [Google Scholar]
- Alabert, C.; Groth, A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol 2012, 13, 153–167. [Google Scholar]
- Li, Q.; Zhou, H.; Wurtele, H.; Davies, B.; Horazdovsky, B.; Verreault, A.; Zhang, Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell 2008, 134, 244–255. [Google Scholar]
- Chen, C.C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008, 134, 231–243. [Google Scholar]
- Battu, A.; Ray, A.; Wani, A.A. ASF1A and ATM regulate H3K56-mediated cell-cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 2011, 39, 7931–7945. [Google Scholar]
- Costelloe, T.; Lowndes, N.F. Chromatin assembly and signalling the end of DNA repair requires acetylation of histone H3 on lysine 56. Subcell. Biochem 2010, 50, 43–54. [Google Scholar]
- Orphanides, G.; LeRoy, G.; Chang, C.H.; Luse, D.S.; Reinberg, D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 1998, 92, 105–116. [Google Scholar]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 400, 284–288. [Google Scholar]
- Winkler, D.D.; Luger, K. The histone chaperone FACT: structural insights and mechanisms for nucleosome reorganization. J. Biol. Chem 2011, 286, 18369–18374. [Google Scholar]
- Keller, D.M.; Zeng, X.; Wang, Y.; Zhang, Q.H.; Kapoor, M.; Shu, H.; Goodman, R.; Lozano, G.; Zhao, Y.; Lu, H. A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol. Cell 2001, 7, 283–292. [Google Scholar]
- Krohn, N.M.; Stemmer, C.; Fojan, P.; Grimm, R.; Grasser, K.D. Protein kinase CK2 phosphorylates the high mobility group domain protein SSRP1, inducing the recognition of UV-damaged DNA. J. Biol. Chem 2003, 278, 12710–12715. [Google Scholar]
- Yarnell, A.T.; Oh, S.; Reinberg, D.; Lippard, S.J. Interaction of FACT, SSRP1, and the high mobility group (HMG) domain of SSRP1 with DNA damaged by the anticancer drug cisplatin. J. Biol. Chem 2001, 276, 25736–25741. [Google Scholar]
- Li, Y.; Keller, D.M.; Scott, J.D.; Lu, H. CK2 phosphorylates SSRP1 and inhibits its DNA-binding activity. J. Biol. Chem 2005, 280, 11869–11875. [Google Scholar]
- Sand-Dejmek, J.; Adelmant, G.; Sobhian, B.; Calkins, A.S.; Marto, J.; Iglehart, D.J.; Lazaro, J.B. Concordant and opposite roles of DNA-PK and the “facilitator of chromatin transcription” (FACT) in DNA repair, apoptosis and necrosis after cisplatin. Mol. Cancer 2011, 10, 74. [Google Scholar]
- Huang, J.Y.; Chen, W.H.; Chang, Y.L.; Wang, H.T.; Chuang, W.T.; Lee, S.C. Modulation of nucleosome-binding activity of FACT by poly(ADP-ribosyl)ation. Nucleic Acids Res 2006, 34, 2398–2407. [Google Scholar]
- Heo, K.; Kim, H.; Choi, S.H.; Choi, J.; Kim, K.; Gu, J.; Lieber, M.R.; Yang, A.S.; An, W. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol. Cell 2008, 30, 86–97. [Google Scholar]
- Siino, J.S.; Nazarov, I.B.; Svetlova, M.P.; Solovjeva, L.V.; Adamson, R.H.; Zalenskaya, I.A.; Yau, P.M.; Bradbury, E.M.; Tomilin, N.V. Photobleaching of GFP-labeled H2AX in chromatin: H2AX has low diffusional mobility in the nucleus. Biochem. Biophys. Res. Commun 2002, 297, 1318–1323. [Google Scholar]
- Kumari, A.; Mazina, O.M.; Shinde, U.; Mazin, A.V.; Lu, H. A role for SSRP1 in recombination-mediated DNA damage response. J. Cell. Biochem 2009, 108, 508–518. [Google Scholar]
- Dejmek, J.; Iglehart, J.D.; Lazaro, J.B. DNA-dependent protein kinase (DNA-PK)-dependent cisplatin-induced loss of nucleolar facilitator of chromatin transcription (FACT) and regulation of cisplatin sensitivity by DNA-PK and FACT. Mol. Cancer Res 2009, 7, 581–591. [Google Scholar]
- Frehlick, L.J.; Eirin-Lopez, J.M.; Ausio, J. New insights into the nucleophosmin/nucleoplasmin family of nuclear chaperones. BioEssays 2007, 29, 49–59. [Google Scholar]
- Laskey, R.A.; Honda, B.M.; Mills, A.D.; Finch, J.T. Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 1978, 275, 416–420. [Google Scholar]
- Kang, Y.J.; Olson, M.O.; Busch, H. Phosphorylation of acid-soluble proteins in isolated nucleoli of Novikoff hepatoma ascites cells. Effects of divalent cations. J. Biol. Chem 1974, 249, 5580–5585. [Google Scholar]
- Okuwaki, M.; Matsumoto, K.; Tsujimoto, M.; Nagata, K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett 2001, 506, 272–276. [Google Scholar]
- Swaminathan, V.; Kishore, A.H.; Febitha, K.K.; Kundu, T.K. Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol. Cell. Biol 2005, 25, 7534–7545. [Google Scholar]
- Koike, A.; Nishikawa, H.; Wu, W.; Okada, Y.; Venkitaraman, A.R.; Ohta, T. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res 2010, 70, 6746–6756. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar]


{kind=link}
{kind=link}
| Remodeler | Activity during NER | Species | Reference |
|---|---|---|---|
| ATP-dependent remodelers | |||
| BRG1 | Interacts with XPC and DDB2 | Mammalian | [4,25] |
| Required for UV survival (extent not determined) | |||
| Snf5 | Required for UV survival | Yeast | [26] |
| Snf6 | Required for UV survival | Yeast | [26,27] |
| Promotes nucleosome rearrangements in response to UV | |||
| Swi2 | Promotes nucleosome rearrangements in response to UV | Yeast | [27] |
| INO80 | Binds DDB1 and is required for recruitment of XPC and XPA | Mammalian | [28] |
| Recruited to UV damage by Rad4 and involved in chromatin restoration after repair | Yeast | [29] | |
| ACF1 | Recruited to UV damage | Mammalian | [30,31] |
| Required for G2/M checkpoint activation upon UV irradiation | |||
| ISW1 | Required for UV survival at high doses | C. elegans | [32] |
| CHD4 | Directly interacts with RNF8 | Mammalian | [33] |
| Histone chaperones | |||
| CAF-1 | Loads newly-synthesized H3-H4 dimers onto chromatin after NER | Mammalian | [34] |
| Asf1 | Transfers H3K56Ac-H4 tetramers to CAF-1 for post-repair deposition | Mammalian | [35] |
| Required for UV survival (extent not determined) | Yeast | [36] | |
| FACT | Directs CK2-dependent p53 phosphorylation upon UV damage | Mammalian | [37] |
| NPM1 | Overexpression promotes UV survival | Mammalian | [38] |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dinant, C.; Bartek, J.; Bekker-Jensen, S. Histone Displacement during Nucleotide Excision Repair. Int. J. Mol. Sci. 2012, 13, 13322-13337. https://doi.org/10.3390/ijms131013322
Dinant C, Bartek J, Bekker-Jensen S. Histone Displacement during Nucleotide Excision Repair. International Journal of Molecular Sciences. 2012; 13(10):13322-13337. https://doi.org/10.3390/ijms131013322
Chicago/Turabian StyleDinant, Christoffel, Jiri Bartek, and Simon Bekker-Jensen. 2012. "Histone Displacement during Nucleotide Excision Repair" International Journal of Molecular Sciences 13, no. 10: 13322-13337. https://doi.org/10.3390/ijms131013322