


Abstract
Cimetidine, an H2 receptor antagonist, has been used to investigate the tubular secretion of organic cations in human kidney. We report a systematic comprehensive analysis of the inhibition potency of cimetidine for the influx and efflux transporters of organic cations [human organic cation transporter 1 (hOCT1) and hOCT2 and human multidrug and toxin extrusion 1 (hMATE1) and hMATE2-K, respectively]. Inhibition constants (Ki) of cimetidine were determined by using five substrates [tetraethylammonium (TEA), metformin, 1-methyl-4-phenylpyridinium, 4-(4-(dimethylamino)styryl)-N-methylpyridinium, and m-iodobenzylguanidine]. They were 95 to 146 μM for hOCT2, providing at most 10% inhibition based on its clinically reported plasma unbound concentrations (3.6–7.8 μM). In contrast, cimetidine is a potent inhibitor of MATE1 and MATE2-K with Ki values (μM) of 1.1 to 3.8 and 2.1 to 6.9, respectively. The same tendency was observed for mouse Oct1 (mOct1), mOct2, and mouse Mate1. Cimetidine showed a negligible effect on the uptake of metformin by mouse kidney slices at 20 μM. Cimetidine was administered to mice by a constant infusion to achieve a plasma unbound concentration of 21.6 μM to examine its effect on the renal disposition of Mate1 probes (metformin, TEA, and cephalexin) in vivo. The kidney- and liver-to-plasma ratios of metformin both were increased 2.4-fold by cimetidine, whereas the renal clearance was not changed. Cimetidine also increased the kidney-to-plasma ratio of TEA and cephalexin 8.0- and 3.3-fold compared with a control and decreased the renal clearance from 49 to 23 and 11 to 6.6 ml/min/kg, respectively. These results suggest that the inhibition of MATEs, but not OCT2, is a likely mechanism underlying the drug-drug interactions with cimetidine in renal elimination.
Introduction
The kidney plays important roles in the elimination of drugs and their metabolites, as well as endogenous waste from circulating blood, and acts as a determinant of systemic drug exposure. Renal elimination of drugs is determined by glomerular filtration, tubular secretion, and reabsorption from the urine. Drug transporters play an indispensable role in the active tubular secretion of drugs in the proximal tubules. For cationic drugs, the basolateral uptake is mediated by OCTs (Oct1 and Oct2 in rodents and OCT2 in humans) and proton/organic cation exchangers, MATEs (Mate1 in rodents and MATE1 and MATE2-K in humans), which are considered to mediate the efflux of cationic drugs into the urine (International Transporter Consortium et al., 2010; Nies et al., 2011; Yonezawa and Inui, 2011). A defect of Mate1 has markedly delayed the systemic elimination of metformin and cephalexin in mice, indicating the importance of Mate1 in the renal elimination of cationic drugs (Tsuda et al., 2009a; Watanabe et al., 2010). We also demonstrated that a potent MATE inhibitor, pyrimethamine, significantly reduced the luminal efflux of TEA and metformin in the liver and kidney in mice (Ito et al., 2010) and the renal clearance of metformin in healthy human subjects (Kusuhara et al., 2011).
Drug-drug interactions (DDIs) involving the inhibition of metabolism and/or excretion prolong the plasma elimination half-lives, leading to the accumulation of victim drugs in the body, and consequently potentiate pharmacological/adverse effects (Kusuhara and Sugiyama, 2008; International Transporter Consortium et al., 2010; Zhang et al., 2011). To avoid severe DDIs in clinical situations, it is now proposed to evaluate the significance of drug transporters on the pharmacokinetics of drug candidates in humans in vivo during drug development. Under such circumstances, there is a great concern regarding drugs that are in vivo inhibitors of drug transporters. Cimetidine, a histamine H2-receptor antagonist, is well known as an inhibitor of the renal organic cation transport system, and it has been used to characterize the elimination pathway of drugs and drug candidates in humans (summarized in Table 1). Cimetidine is now recommended as an inhibitor to evaluate the impact of OCT2 on the renal elimination of drug candidates when they are substrates of OCT2, and their renal elimination accounts for more than 50% of systemic elimination (International Transporter Consortium et al., 2010). However, whether the inhibition of OCT2 by cimetidine has in vivo relevance remains controversial. The reported inhibition constants (Ki) of cimetidine vary greatly depending on the article, ranging from 11 to 1650 μM (Supplemental Table 1). This difference is partly attributable to the substrate tested and experimental system used. Taking the lowest value obtained using cationic styryl dye [4-(4-(dimethylamino)styryl)-N-methylpyridinium (ASP+)] as a substrate, the inhibition of OCT2 by cimetidine becomes significant at clinical doses (Pietig et al., 2001). The median reported Ki for cimetidine is 120 μM, far greater than the unbound concentration of cimetidine at clinical doses, indicating the unlikelihood of OCT2 inhibition by cimetidine at therapeutic concentrations. In contrast, because the MATE1 Ki for cimetidine is similar to or somewhat lower than the unbound concentration, it is proposed that a pharmacokinetic interaction in the renal elimination of organic cations with cimetidine is caused by an inhibition of MATE1 (Matsushima et al., 2009; Tsuda et al., 2009b).
Pharmacokinetic drug-drug interactions caused by cimetidine in the tubular secretion
All of the renal clearance was significantly decreased by coadministration of cimetidine. Each value represents the mean ± S.D. (mean ± S.E.a).
The purpose of this study was to conduct a comprehensive in vitro inhibition study to compare the inhibition potency of cimetidine for OCT2 and MATEs in humans and mice and elucidate the transporter responsible for the interaction. Inhibition constants of cimetidine for human OCT2, MATE1, and MATE2-K were determined by using five known substrates, including ASP+, which has provided the lowest OCT2 Ki for cimetidine reported in the literature. Furthermore, we also performed an in vitro inhibition study of the murine organic cation transporters (Oct1, Oct2, and Mate1) and an in vivo pharmacokinetic interaction study in mice to obtain insight into the mechanism of DDIs caused by cimetidine. This study contributes to the understanding of pharmacokinetic interaction studies using cimetidine as an inhibitor.
Materials and Methods
Materials.
[14C]TEA (3.2 mCi/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA), [14C]metformin (26 mCi/mmol) was purchased from Moravec Biochemicals (Brea, CA), and [3H]1-methyl-4-phenylpyridinium (MPP+) (80 Ci/mmol) was purchased from American Radiochemical Company (St. Louis, MO). Unlabeled metformin and m-iodobenzylguanidine (MIBG) were purchased from Sigma (St. Louis, MO), and ASP was purchased from Invitrogen (Carlsbad, CA). All other chemicals used were commercially available and of analytical grade.
Animals.
Male ddY mice were purchased from Japan SLC (Shizuoka, Japan). All animals were maintained under standard conditions with a reverse dark-light cycle and kept for at least 7 days before pharmacological experiments. Food and water were available ad libitum. The mice used in the present study were from 8 to 9 weeks old. The studies were conducted in accordance with the guidelines provided by the Institutional Animal Care Committee at the Graduate School of Pharmaceutical Sciences, University of Tokyo, Tokyo, Japan.
In Vitro Transport Study Using cDNA Transfectants.
hOCT1-HEK293, hOCT2-HEK293, mOct1-HEK293, mOct2-HEK293, hMATE1-HEK293, hMATE2-K-HEK293, and mMate1-HEK293 were constructed previously (Busch et al., 1998; Müller et al., 2005; Matsushima et al., 2009; Ito et al., 2010). Cells were seeded 72 h before the transport assay in poly-l-lysine- and poly-l-ornithine-coated 12-well plates at a density of 1.5 × 105 cells per well. For the transport study, the cell culture medium was replaced with culture medium supplemented with 5 mM sodium butyrate 24 h before the transport assay to induce expression of the transporters. The transport study was conducted as described previously (Ito et al., 2010). Uptake was initiated by the addition of substrates after the cells had been washed twice and preincubated with Krebs-Henseleit buffer at 37°C for 15 min. The Krebs-Henseleit buffer consisted of 118 mM NaCl, 23.8 mM NaHCO3, 4.8 mM KCl, 1.0 mM KH2PO4, 1.2 mM MgSO4, 12.5 mM HEPES, 5.0 mM glucose, and 1.5 mM CaCl2 and was adjusted to pH 7.4. Uptake was terminated at the designated times by the addition of ice-cold Krebs-Henseleit buffer after the removal of the incubation buffer. The cells were solubilized with NaOH overnight at 4°C, and then neutralized with HCl. To measure ASP and MIBG, aliquots were used for LC-MS/MS quantification as described below. The radioactivity of radiolabeled compounds in aliquots was measured by liquid scintillation counting. The protein concentration was determined by using the Lowry method with bovine serum albumin as a protein standard as described previously (Lowry et al., 1951).
Transport Study Using Kidney Slices.
Uptake studies were carried out as described previously (Ito et al., 2010). Slices (300 μm in thickness) of whole kidneys from male ddY mice were kept in ice-cold oxygenated buffer before use. The buffer for the present study consisted of 120 mM NaCl, 16.2 mM KCl, 1 mM CaCl2, 1.2 mM MgSO4, and 10 mM NaH2PO4/Na2HPO4 adjusted to pH 7.5. Two slices were selected and then incubated at 37°C on a 12-well plate with 1 ml of oxygenated buffer containing 3.8 μM [14C]metformin in each well after preincubation of slices for 5 min at 37°C. After incubation for 10 min, slices were rapidly removed from the incubation buffer, washed twice with ice-cold buffer, blotted on filter paper, weighed, and dissolved in 1 ml of Soluene-350 (PerkinElmer Life and Analytical Sciences) at 55°C for 12 h. The radioactivity in the specimens was determined in a scintillation counter after adding scintillation cocktail (Hionic Fluor; PerkinElmer Life and Analytical Sciences). The uptake activity of Oct1 and Oct2 in the mouse kidney slices was checked with TEA as the positive control.
Steady-State Infusion Study of TEA, Metformin, and Cephalexin in Mice Treated With and Without Cimetidine.
After anesthesia with isoflurane, the urinary bladder was catheterized. TEA (128 nmol/min/kg), metformin (100 nmol/min/kg), and cephalexin (10 nmol/min/kg) were infused via the jugular vein. In the cimetidine-treated group, cimetidine (1500 nmol/min/kg) was simultaneously infused continuously via the jugular vein. Blood samples were collected via the jugular vein at 50, 70, and 90 min for TEA or 60, 90, and 120 min for metformin and cephalexin after administration and centrifuged to obtain plasma. Urine specimens were collected at 50 to 70 and 70 to 90 min for TEA or 60 to 90 and 90 to 120 min for metformin and cephalexin. At the end of the experiment, the kidneys and liver were removed. Drug concentration was determined by liquid scintillation counting for TEA and LC-MS analysis for metformin and cephalexin.
Quantification of Drug Concentrations by LC-MS/MS.
The kidney and liver were homogenized in a 4-fold volume of phosphate-buffered saline. The urine specimens were diluted with a 50-fold volume of water. In the case of cimetidine, all specimens were prepared by an additional 10-fold dilution. All biological specimens were mixed with a 2-fold volume of acetonitrile. Mixed solutions were centrifuged at 20,000g for 10 min. The supernatants were mixed with a 4-fold volume of water and centrifuged at 20,000g for 10 min, and an aliquot of the supernatant was subjected to LC-MS analysis. The aliquots (20 μl) obtained from the uptake study were precipitated with 80 μl of acetonitrile. Mixed solutions were centrifuged at 20,000g for 10 min. The supernatants were mixed with a 4-fold volume of water. Mixed solutions were then centrifuged at 20,000g for 10 min to remove particulates, and an aliquot of the supernatants was used for LC-MS/MS analysis.
An AB SCIEX QTRAP 5500 mass spectrometer (Applied Biosystems/MDS Sciex, Foster City, CA) equipped with a Prominence LC system (Shimadzu, Kyoto, Japan), operated in the electron spray ionization mode, was used for the analysis of cephalexin, ASP, and MIBG. An LCMS-2010EV system equipped with a Prominence LC system (Shimadzu) was used for the analysis of metformin and cimetidine. Detailed LC conditions and mass-to-charge ratios are shown in Supplemental Table 2.
Kinetic Analyses.
Specific uptake was calculated by subtracting the uptake by the vector-transfected cells from the uptake by the cDNA-transfected cells. Kinetic parameters were calculated by using the following equations:

 where CLuptake (+Inhibitor) and CLuptake (control) are the uptake clearance determined in the presence or absence of the inhibitor, respectively, I is the concentration of the inhibitor, Ki is the inhibition constant, Pdif is the nonsaturable uptake clearance, v is the uptake velocity of the substrate, Vmax is the maximum uptake rate, Km is the Michaelis constant, and S is the substrate concentration in the medium. Fitting was performed with a nonlinear least-squares method using the MULTI program (Yamaoka et al., 1981), and the Damping Gauss-Newton algorithm was used for curve fitting.
where CLuptake (+Inhibitor) and CLuptake (control) are the uptake clearance determined in the presence or absence of the inhibitor, respectively, I is the concentration of the inhibitor, Ki is the inhibition constant, Pdif is the nonsaturable uptake clearance, v is the uptake velocity of the substrate, Vmax is the maximum uptake rate, Km is the Michaelis constant, and S is the substrate concentration in the medium. Fitting was performed with a nonlinear least-squares method using the MULTI program (Yamaoka et al., 1981), and the Damping Gauss-Newton algorithm was used for curve fitting.
Pharmacokinetic Analysis.
The fractional urinary excretion ratio (Furine), total body clearance (CLtot), and renal clearance (CLR) were calculated by using the following equations:


 where V and I represent urinary excretion rate and infusion rate, respectively. Dose and X represent the amount of ligand administered in 1 h and excreted into urine from 30 to 90 min for TEA or 60 to 120 min for metformin and cephalexin. AUCp represents the area under the time-plasma concentration curve for ligands from 30 to 90 min for TEA or 60 to 120 min for metformin and cephalexin. The apparent tissue-to-plasma concentration ratio (Kp, tissue) was calculated by using the following equation:
where V and I represent urinary excretion rate and infusion rate, respectively. Dose and X represent the amount of ligand administered in 1 h and excreted into urine from 30 to 90 min for TEA or 60 to 120 min for metformin and cephalexin. AUCp represents the area under the time-plasma concentration curve for ligands from 30 to 90 min for TEA or 60 to 120 min for metformin and cephalexin. The apparent tissue-to-plasma concentration ratio (Kp, tissue) was calculated by using the following equation:
 where Ctissue and Cp represent tissue and plasma concentrations of ligands, respectively, at 90 or 120 min after administration.
where Ctissue and Cp represent tissue and plasma concentrations of ligands, respectively, at 90 or 120 min after administration.
Statistical Analysis.
Data are presented as the mean ± S.E. Student's two-tailed unpaired t test and a one-way analysis of variance followed by Dunnett's post hoc test were used to identify significant differences between groups where appropriate. p < 0.05 was considered to be statistically significant.
Results
In Vitro Inhibition of hOCT1, hOCT2, hMATE1, and hMATE2-K in cDNA-Transfected Cells.
The uptake of the five compounds tested was significantly greater in HEK293 cells expressing hOCT1, hOCT2, hMATE1, or hMATE2-K (Fig. 1). The subsequent analyses were conducted for the uptake determined at times summarized in Supplemental Table 3. MIBG showed somewhat high transport activity by hOCT1 followed by ASP and MPP+, whereas the transport activity of MIBG by hOCT2 was not as high as ASP and MPP+ and similar to those of metformin and TEA. hMATE1 and hMATE2-K showed similar profiles except MIBG, the transport activity of which was higher than that of ASP for hMATE1, but vice versa for hMATE2-K.
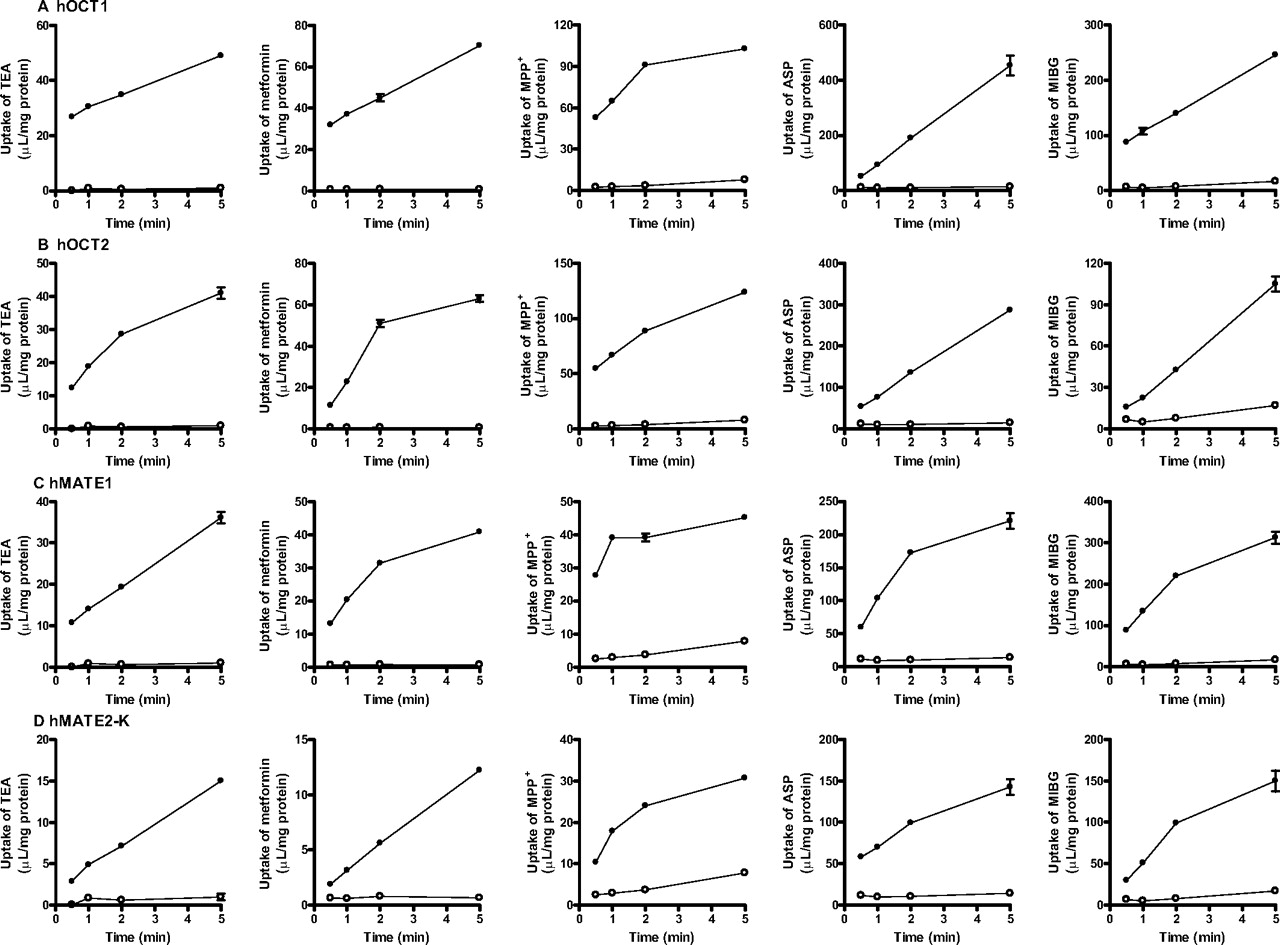
Time profiles of cationic compounds uptake by hOCT1 (A), hOCT2 (B), hMATE1 (C), and hMATE2-K (D). Time-dependent uptake of TEA (31 μM), metformin (3.8 μM), MPP+ (100 nM), ASP (1.0 μM), or MIBG (1.0 μM) were determined in HEK293 cells expressing transporter-expressing cells (●) and mock cells (○) at 37°C. Each point represents the mean value, and error bars represent the S.E. (n = 3).
Cimetidine significantly inhibited the uptake by all of the transporters tested in a concentration-dependent manner (Fig. 2). The cimetidine Ki values are summarized in Table 2. There was no marked substrate dependence in cimetidine Ki values for hOCT1 and hOCT2. In contrast, cimetidine Ki values for both hMATE1 and hMATE2-K showed some substrate dependence; the difference in Ki was the largest when TEA and metformin were used as substrates (3-fold). All of the Ki values for hMATE1 and hMATE2-K were markedly smaller than those for hOCT2, although the difference became smaller when metformin was used as substrate compared with the other substrates (Fig. 2). Kinetic parameters were determined in the absence and presence of cimetidine (100 μM for hOCT1 and hOCT2 and 3 μM for hMATE1 and hMATE2-K) to examine the type of inhibition by cimetidine (Fig. 3). The Km was apparently increased in the presence of cimetidine, without affecting Vmax, indicating competitive inhibition.

Effect of cimetidine on the uptake of cationic compounds by hOCT1 (○), hOCT2 (□), hMATE1 (●), and hMATE2-K (■). HEK293 cells expressing hOCT1, hOCT2, hMATE1, or hMATE2-K were incubated with TEA (31 μM), metformin (3.8 μM), MPP+ (100 nM), ASP (1.0 μM), or MIBG (1.0 μM) at 37°C in the presence of cimetidine at the designated concentrations. The specific uptake of substrates was calculated by subtracting the uptake by mock cells from the uptake by cDNA transfectants. The solid lines represent the fitted line obtained by nonlinear regression analysis based on eq. 1 as described under Materials and Methods. Each point represents the mean value, and error bars represent the S.E. (n = 3).
Cimetidine inhibition constants for the renal organic cation transporters
Data shown in Fig. 2 and 5 were used to determine these Ki values calculated by using nonlinear regression analysis as described under Materials and Methods. Each parameter represents the mean value ± computer-calculated S.D.

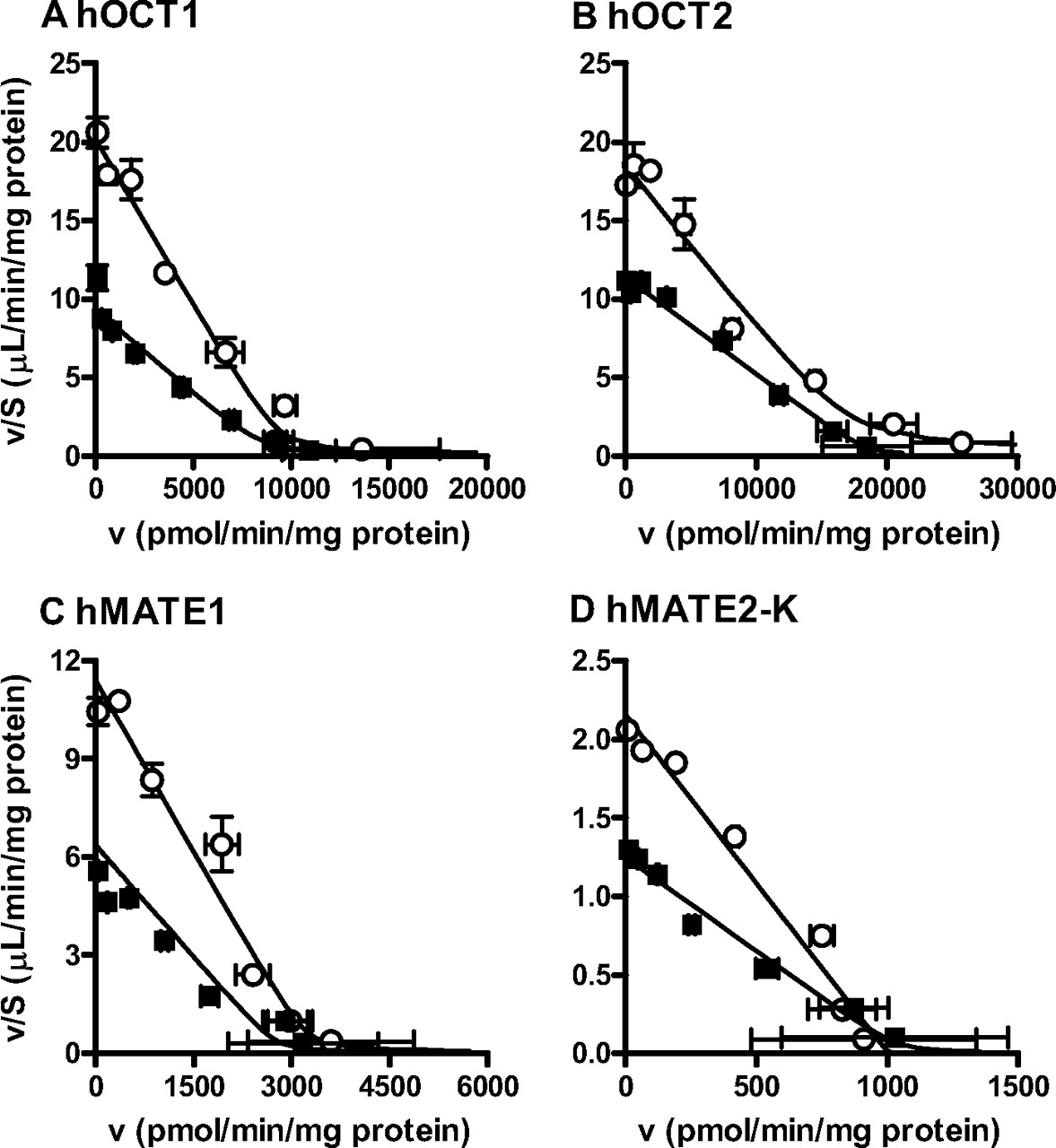
Type of cimetidine inhibition for hOCT1 (A), hOCT2 (B), hMATE1 (C), and hMATE2-K (D). HEK293 cells expressing hOCT1, hOCT2, hMATE1, or hMATE2-K were incubated at 37°C with various concentrations of metformin (4–30,000 μM) in the presence (■) or absence (○) of cimetidine (100 μM for hOCT1 and hOCT2 and 3 μM for hMATE1 and hMATE2-K). Concentration dependence of the transport activities is shown as an Eadie-Hofstee plot. The solid lines represent the fitted line obtained by nonlinear regression analysis based on eq. 2 as described under Materials and Methods. Each point represents the mean value, and error bars represent the S.E. (n = 3).
In Vitro Inhibition of mOct1, mOct2, and mMate1 in cDNA Transfectants and Metformin Uptake by Mouse Kidney Slices.
The uptake of the five compounds tested was significantly greater in HEK293 cells expressing mOct1, mOct2, and mMATE1 (Fig. 4). The subsequent analyses were conducted for the uptake determined at the times summarized in Supplemental Table 3. Unlike hOCT1, the transport activities of ASP and MIBG by mOct1 were not as high as MPP+ and similar to that of TEA. Metformin transport activity by mOct1 was the lowest among the substrates tested. mOct2 was characterized by low transport activity of ASP compared with hOCT2. mMate1 showed a similar profile to hMATE1 and hMATE2-K.

Time profiles of cationic compounds uptake by mOct1 (A), mOct2 (B), and mMate1 (C). Time-dependent uptake of TEA (31 μM), metformin (3.8 μM), MPP+ (100 nM), ASP (1.0 μM), or MIBG (1.0 μM) was determined in HEK293 cells expressing transporter-expressing cells (mOct1, mOct2, or mMate1; ●) and mock cells (○) at 37°C. Each point represents the mean value, and error bars represent the S.E. (n = 3).
Cimetidine significantly inhibited the uptake by all of the transporters tested in a concentration-dependent manner (Fig. 5). The Ki values are summarized in Table 2. The cimetidine Ki values for mOct1 were somewhat smaller than those for hOCT1, particularly, and those for MPP+ uptake showed 4-fold smaller values for mOct1 than for hOCT1. The cimetidine Ki values for mOct1, mOct2, and mMate1 were similar to their corresponding human isoforms. Kinetic parameters were determined in the absence and presence of cimetidine (100 μM for mOct1 and mOct2 and 3 μM for mMate1) to examine the type of inhibition by cimetidine (Fig. 6). The Km was apparently increased in the presence of cimetidine, without affecting Vmax, indicating competitive inhibition. The effect of cimetidine on metformin uptake by mouse kidney slices is shown in Fig. 7. Cimetidine significantly inhibited the uptake of metformin at 200 and 2000 μM.

Effect of cimetidine on the uptake of cationic compounds by mOct1 (○), mOct2 (□), and mMate1 (●). HEK293 cells stably expressing mOct1, mOct2, or mMate1 were incubated with TEA (31 μM), metformin (3.8 μM), MPP+ (100 nM), ASP (1.0 μM), or MIBG (1.0 μM) at 37°C in the presence of cimetidine at the designated concentrations. The specific uptake of substrates was calculated by subtracting the uptake by mock cells from the uptake by cDNA transfectants. The solid lines represent the fitted line obtained by nonlinear regression analysis based on eq. 1 as described under Materials and Methods. Each point represents the mean value, and error bars represent the S.E. (n = 3).

Type of cimetidine inhibition on the uptake of metformin by mOct1 (A), mOct2 (B), and mMate1 (C). HEK293 cells expressing mOct1, mOct2, or mMate1 were incubated at 37°C with various concentrations of metformin (4–30,000 μM) in the presence (■) or absence (○) of cimetidine (100 μM for mOct1 and mOct2 and 3 μM for mMate1). Concentration dependence of the transport activities is shown as an Eadie-Hofstee plot. The solid lines represent the fitted line obtained by nonlinear regression analysis based on eq. 2 as described under Materials and Methods. Each point represents the mean value, and error bars represent the S.E. (n = 3).

Effect of cimetidine on metformin uptake by kidney slices. The inhibitory effect of cimetidine was determined by using kidney slices. The uptake of [14C]metformin (0.1 μCi/ml, 3.8 μM) was measured for 10 min at 37°C. The uptake of [14C]metformin was determined in the presence of various concentrations of cimetidine (20–2000 μM). Each point represents the mean value, and error bars represent the S.E. (n = 3). **, p < 0.01.
Effect of Cimetidine on the Renal Elimination and Kidney Concentration of TEA, Metformin, and Cephalexin.
Cimetidine was administered to mice by a constant infusion. The plasma concentration of cimetidine reached a plateau 60 min after the start of infusion (26.7 ± 1.0 μM). Taking the unbound fraction in the plasma into consideration (Kalvass et al., 2007), the unbound cimetidine concentration in the plasma was 21.6 μM.
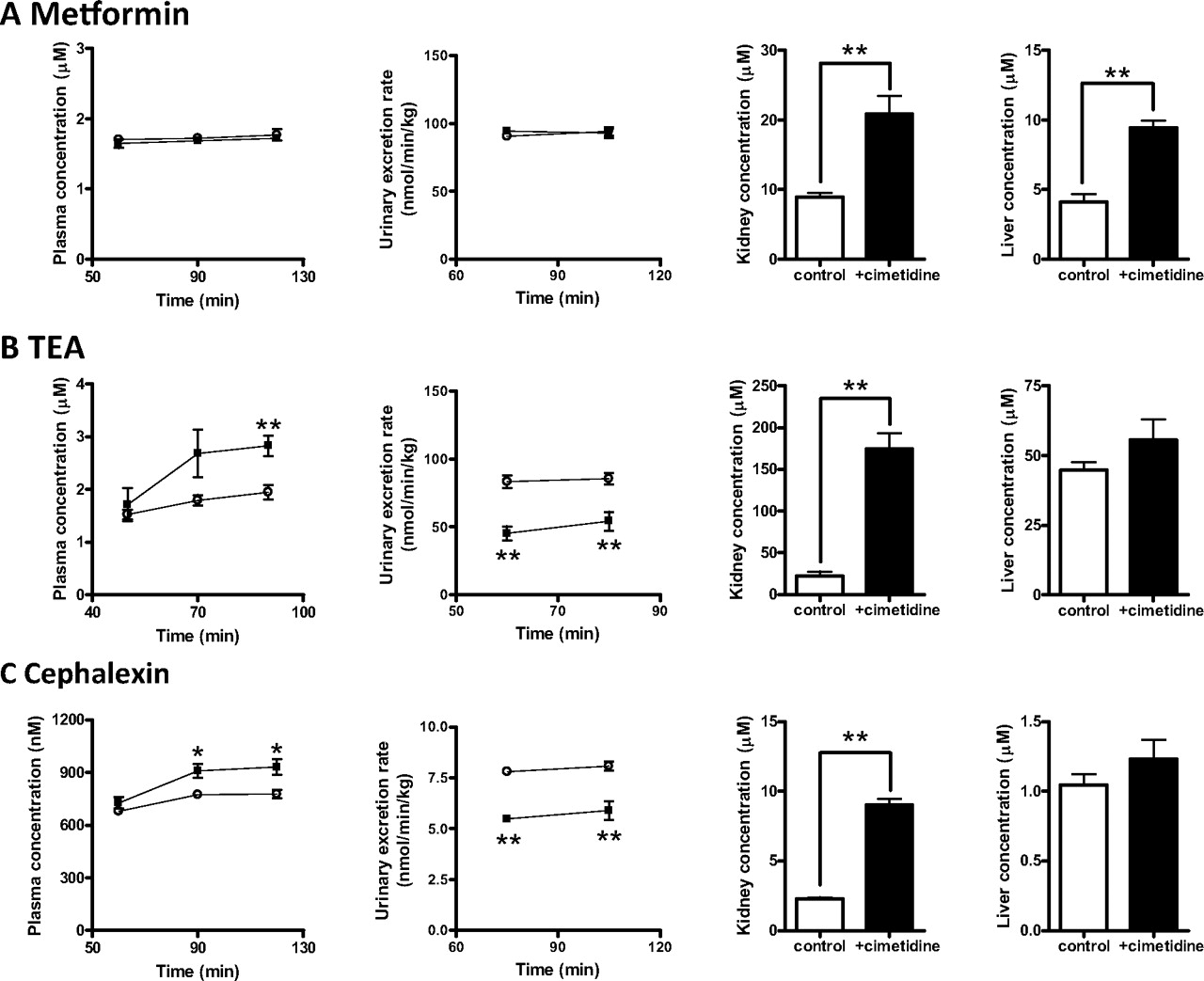
TEA, metformin, and cephalexin were administered to mice by intravenous infusion. The concentrations of drugs in the plasma, kidney, and liver, as well as the urinary excretion rate, were measured under steady-state conditions with and without the coinfusion of cimetidine. Cimetidine treatment significantly increased the concentrations of metformin in the kidney and liver, whereas it affected neither plasma concentration nor urinary excretion rates (Fig. 8A). The renal clearance of metformin with regard to the plasma concentration was not changed by cimetidine, and both Kp, kidney and Kp, liver were significantly elevated in mice treated with cimetidine (Table 3). Cimetidine treatment significantly increased the plasma and kidney concentration of TEA and cephalexin and decreased the urinary excretion rate (Fig. 8, B and C). The total body clearance and renal clearance of TEA and cephalexin with regard to the plasma concentration were significantly decreased by cimetidine, and Kp, kidney, but not Kp, liver, was significantly elevated in mice treated with cimetidine (Table 3). The nonrenal clearance of TEA and cephalexin was greater in mice treated with cimetidine (25 versus 33 ml/min/kg for TEA, p < 0.05); 2.7 versus 5.0 ml/min/kg for cephalexin, p < 0.001).

Effect of cimetidine on the renal elimination of metformin (A), TEA (B), and cephalexin (C) in mice. Cimetidine was administered to male ddY mice by intravenous infusion (1500 nmol/min/kg) simultaneously with TEA (128 nmol/min/kg), metformin (100 nmol/min/kg), or cephalexin (10 nmol/min/kg). Blood samples were collected at the designated times, and urine specimens were collected at 20-min intervals from 50 to 90 min for TEA or 30-min intervals from 60 to 120 min for metformin and cephalexin. At the end of the experiment, the kidneys and liver were removed. Drug concentrations in the plasma, urine, kidney, and liver were determined as follows: TEA was quantified by liquid scintillation counting, Metformin was quantified by LC-MS, and Cephalexin was quantified by LC-MS/MS. Each point represents the mean value, and error bars represent the SE (n = 6). *, p < 0.05; **, p < 0.01.
Pharmacokinetic parameters for TEA, metformin, and cephalexin in control and cimetidine-treated mice
Each value was determined from the data shown in Fig. 8. The equations to determine the kinetic parameters are described under Materials and Methods. Each value represents the mean ± S.E. (n = 6).
Discussion
A comprehensive in vitro inhibition study was conducted to examine whether the cimetidine Ki for renal organic cation transporters is substrate-dependent in cDNA-transfected cells, and a pharmacokinetic interaction study was conducted in mice to obtain further insight into the mechanism of DDI.
Consistent with the fact that cimetidine is a substrate of renal organic cation transporters (Dudley et al., 2000; Ito et al., 2010), cimetidine acts as a competitive inhibitor of other substrates (Fig. 3). Unexpectedly, the low cimetidine Ki for the uptake of ASP by hOCT2 could not be reproduced in our study. The cimetidine Ki values for hOCT2 were similar irrespective of the substrates, and they were higher than the unbound concentration of cimetidine in the plasma at clinical doses, indicating that inhibition of OCT2 by clinical doses of cimetidine is negligible. Among the reports, relatively large Ki of cimetidine reported by Umehara et al. (2007) might have been the result of difference in OCT2 variants (NM_153191), because OCT2-A, which is an alternatively spliced variant of OCT2, lacking an approximately 60-amino acid residue sequence at the C terminus, was used in the inhibition study. In fact, Urakami et al. (2002) reported a difference in the inhibition potency of cimetidine for hOCT2 and hOCT2-A. In addition, we speculate that quantification of the amount of test compounds by fluorescence microscopy may overestimate the inhibition potency for cimetidine. Biermann et al. (2006) and Pietig et al. (2001) reported relatively small Ki values for cimetidine (Supplemental Table 1). They quantified cellular accumulation of ASP and amiloride by using fluorescence microscopy. The possibility that cimetidine is a more potent inhibitor for the uptake of organic cations in the kidney than in cDNA transfectants can be excluded by an in vitro inhibition study using mouse kidney slices. The inhibition potency of cimetidine in mouse kidney slices was comparable with its inhibition potency in mOcts-HEK; cimetidine did not affect the metformin uptake at 20 μM, but did so moderately at 200 μM (Fig. 7).
On the other hand, consistent with previous reports (Matsushima et al., 2009; Tsuda et al., 2009b), cimetidine is a potent competitive inhibitor of hMATE1. The Ki value of cimetidine for hMATE2-K was somewhat larger than that for hMATE1 (Table 2), but the difference was not convincing compared with the previous reported values (6.6-fold; Tsuda et al., 2009b). The reason for this discrepancy remains unknown, because there is no crucial difference in the MATE1 and MATE2-K isoforms or the host cells used. Even the largest Ki values for hMATE1 and hMATE2-K, which were determined by using metformin, were 25- and 14-fold smaller than those for hOCT2. Thus, hMATE1 and hMATE2-K were more susceptible to cimetidine than hOCT2 in vitro. Assuming that the unbound concentration of cimetidine in the kidney is similar to that in the plasma (3.6–7.8 μM), it is predicted that hMATE1 and hMATE2-K are decreased to 12 to 51 and 21 to 66% of the controls, respectively, and consequently, the tubular secretion will be decreased to 50% of the control or less unless uptake is the rate-determining process.
To obtain an insight into the DDI mechanism, a pharmacokinetic interaction study was conducted in mice. In mice proximal tubules, both Oct1 and Oct2 mediate the basolateral uptake of organic cations in the kidney (Jonker et al., 2003), whereas Mate1 mediates the luminal efflux into the urine. The cimetidine Ki for mOct1 was slightly smaller than that for mOct2; in particular, it was 3-fold smaller when MPP+ was used as substrate. However, they are 15-fold larger than the Ki for mMate1 (Table 2). Consistent with human data, mMate1 is more susceptible to cimetidine than mOct1 and mOct2. The in vivo experiment was designed to produce an unbound cimetidine concentration in the plasma that was similar to that in humans at clinical doses, which is firmly below its Ki values for mOcts, but above the Ki for mMate1. In fact, cimetidine could not inhibit the uptake of metformin by mouse kidney slices at the concentration relevant to the in vivo study (Fig. 7). TEA, metformin, and cephalexin were selected as test drugs, the tubular secretion of which is mediated by Mate1 (Tsuda et al., 2009b; Ito et al., 2010; Watanabe et al., 2010). Cimetidine significantly reduced the renal clearance of TEA and cephalexin with regard to the plasma concentration, but not metformin, and it increased the kidney-to-plasma ratio of TEA, metformin, and cephalexin (Table 3).
Based on the in vivo data of TEA in Oct1(−/−), Oct2(−/−), and Oct1/2(−/−) mice (Jonker et al., 2003), inhibition of the basolateral uptake should reduce the kidney-to-plasma ratio. Thus, the luminal efflux of TEA, metformin, and cephalexin is inhibited by cimetidine, resulting in the reduced renal clearance of TEA and cephalexin with regard to the plasma concentration. It must be noted that the kidney-to-plasma ratio in Table 3, which is the product of the availability in the kidney and true kidney-to-plasma ratio (the ratio of the concentration in the tissue and capillary) is an apparent parameter. Therefore, the effect of cimetidine on the kidney-to-plasma ratio of TEA is partly attributable to higher availability in the kidney. Absence of an effect of cimetidine on the plasma concentration of metformin is attributable to the blood flow-limited renal elimination and the magnitude of the luminal efflux relative to the basolateral efflux as we reported previously (Ito et al., 2010). The results support our hypothesis that DDIs caused by cimetidine during renal elimination involve inhibition of MATEs, at least partly. It is worth mentioning that cimetidine also significantly increased the liver-to-plasma ratio of metformin (Table 3). Because we reported that inhibition of mMate1-mediated canalicular efflux results in a significant increase in the ratio (Ito et al., 2010), cimetidine probably inhibits the canalicular efflux mediated by mMate1. A pharmacokinetic interaction between metformin and cimetidine enhances the pharmacological effect of metformin on the liver, inhibition of gluconeogenesis from lactate in healthy subjects (Somogyi et al., 1987). The clinical DDI may also involve the inhibition of canalicular efflux in humans. On the other hand, unlike kidney, the liver concentration of TEA and cephalexin was unchanged by cimetidine although they should be taken up by hepatocytes, considering its distribution volume in the liver above the extracellular space. TEA and cephalexin may undergo canalicular efflux by other transporters, or canalicular efflux is not the major elimination pathway from the liver for these two compounds.
In contrast to long-held beliefs, the present study excludes the possibility of competitive inhibition of OCT2 as the mechanism underlying the DDI caused by cimetidine. Currently, inhibition of MATEs seems the most likely mechanism. The effect of cimetidine has been examined after repeated doses for a couple of days; however, a single dose of cimetidine is sufficient to achieve plasma concentrations capable of inhibiting MATEs. Indeed, a single dose of cimetidine caused significant inhibition of the renal elimination of procainamide (Table 1). Previous clinical pharmacokinetic interaction studies, summarized in Table 1, suggest the importance of MATEs as the luminal efflux transporters for organic cations in humans. Actually, of the drugs listed in Table 1, cephalexin, fexofenadine, levofloxacin, metformin, nicotine, and procainamide are found to be MATE substrates in vitro. Wang et al. (2008) reported that the effect of cimetidine on the renal clearance of metformin depends on the OCT2 genotypes. A single-nucleotide polymorphism at the 808 nucleotide position (rs316019), substituting the amino acid from alanine to serine, is associated with the effect of cimetidine on the renal clearance of metformin as well as metformin clearance. The effect of cimetidine was very weak in the genotype of TT compared with other genotypes, although metformin undergoes significant tubular secretion even in the genotype. Substitution of the amino acid relieves the inhibitory effect of cimetidine in vitro (Zolk et al., 2009); however, the Ki remained greater than the unbound concentration of cimetidine. The single-nucleotide polymorphism may be associated with a lower kidney concentration of cimetidine, resulting in attenuation of its inhibitory effect on MATEs. There is another possibility that cimetidine inhibits OCT2 function by mechanisms other than competitive inhibition, because the study was designed to involve repeated administration of cimetidine for 2 to 12 days. Direct measurement of the uptake clearance before and after cimetidine treatment using imaging technologies for evaluating tissue concentration will be necessary. MIBG, a single-photon emission computed tomographic ligand, is also a substrate of hOCT2 and hMATEs (Fig. 1), and together with the fact that the renal elimination of MIBG involves tubular secretion in healthy subjects (Blake et al., 1989), a pharmacokinetic interaction study using single-photon emission computed tomography may provide a definitive explanation of the involvement of OCT2 in the DDI caused by cimetidine.
In conclusion, competitive inhibition of OCT2 is unlikely as a mechanism underlying the DDI caused by cimetidine in the renal elimination of cationic drugs. Competitive inhibition of MATEs by cimetidine is likely to be important in vivo at its clinical doses. Cimetidine, in addition to pyrimethamine, can be used as an in vivo inhibitor of hMATEs.
Authorship Contributions
Participated in research design: Ito, Kusuhara, and Sugiyama.
Conducted experiments: Ito, Yokochi, and Toyoshima.
Contributed new reagents or analytic tools: Inoue and Yuasa.
Performed data analysis: Ito and Kusuhara.
Wrote or contributed to the writing of the manuscript: Ito, Kusuhara, and Sugiyama.
Footnotes
This study was supported in part by the Japan Society for the Promotion of Science [Grant-in-Aid for Scientific Research (A) 20249008 (to Y.S.) and Grant-in-Aid for Scientific Research (B) 23390034 (to H.K.)]; and Health and Labor Sciences Research Grants (Research on Regulatory Science of Pharmaceuticals and Medical Devices) (to Y.S.).
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
↵
 The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- OCT
- organic cation transporter
- hOCT
- human OCT
- mOct
- mouse OCT
- ASP
- 4-(4-(dimethylamino)styryl)-N-methylpyridinium
- DDI
- drug-drug interaction
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MATE
- multidrug and toxin extrusion
- hMATE
- human MATE
- mMate
- mouse MATE
- MIBG
- m-iodobenzylguanidine
- MPP+
- 1-methyl-4-phenylpyridinium
- TEA
- tetraethylammonium
- HEK
- human embryonic kidney
- CL
- clearance.

- Received June 11, 2011.
- Accepted November 8, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}