Article Text

Abstract
Introduction Antioxidants may have positive impact on diabetic polyneuropathy (DPN), presumably due to alleviation of oxidative stress. We aimed to evaluate the efficacy and safety of combination of antioxidants: succinic acid, inosine, nicotinamide, and riboflavin (SINR) in the treatment of DPN.
Research design and methods In a double-blind, placebo-controlled clinical trial, men and women aged 45–74 years with type 2 diabetes and symptomatic DPN, with initial Total Symptom Score (TSS) ˃5, were randomized into experimental (n=109) or placebo (n=107) group. Patients received study medication/placebo intravenously for 10 days, followed by oral administration for 75 days. Statistical significance was defined as a two-tailed p<0.05.
Results In SINR group, mean TSS change after 12 weeks was –2.65 (±1.46) vs –1.73 (±1.51) in the placebo group (p<0.0001; t-test). Reduction of symptoms in the SINR group was achieved regardless of hemoglobin A1c levels, but better results were observed in patients with initial TSS <7.5. The analysis of TSS subscores revealed statistically significant between-group differences by dynamics of the intensity of paresthesia and of numbness starting from day 11 (p=0.035 and p=0.001, respectively; mixed model); by day 57, statistically significant between-group differences were detected also by dynamics of burning intensity (p=0.005; mixed model). Study limitations are small effect size, moderate proportion of patients with severe DPN symptoms, subjective assessment of outcomes, exclusion of participants who received injectable glucose-lowering medications other than insulins, and patients with uncontrolled and type 1 diabetes.
Conclusions The combination of SINR effectively alleviates DPN symptoms in patients with type 2 diabetes.
Trial registration number ClinicalTrials.gov Registry (NCT04649203; Unique Protocol ID: CTF-III-DM-2019).
- Diabetes Complications
- Diabetes Mellitus, Type 2
- Diabetic Neuropathies
Data availability statement
Data may be obtained from a third party and are not publicly available. Data were collected and stored by an independent CRO.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Diabetic polyneuropathy causes troublesome symptoms and may negatively affect the quality of life in patients with type 2 diabetes mellitus. Some studies have shown that antioxidants (eg, alpha-lipoic acid) may alleviate symptoms of diabetic polyneuropathy.
WHAT THIS STUDY ADDS
A randomized double-blind placebo-controlled study has demonstrated that treatment with combination of antioxidants (succinic acid, inosine, nicotinamide, and riboflavin) for 3 months helps to reduce paresthesia, numbness and burning in patients with diabetic polyneuropathy. This effect was achieved regardless of the degree of compensation for type 2 diabetes, but was associated with less severe initial symptoms.
Treatment with combination of succinic acid, inosine, nicotinamide, and riboflavin, administered together with other treatments of type 2 diabetes, neither raised safety concerns nor induced hypoglycemia episodes.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Combination of succinic acid, inosine, nicotinamide, and riboflavin is a new and safe treatment, which may be offered to patients with mild and moderate numbness, paresthesias and burning resulting from diabetic polyneuropathy, in addition to standard therapy in order to reduce symptoms.
Introduction
The reported prevalence of diabetic polyneuropathy (DPN) varies from 6% to 51% in adult patients;1 its detection is increased to 90% with the use of electrophysiological methods.2 3 Symptoms of DPN, such as neuropathic pain and decreased sensation, can cause falls and fractures, impair quality of life, restrict activities of daily living, provoke depressive symptoms, and contribute to formation of foot complications.4–6 Treatment options for DPN which effectively target the underlying nerve damage are still lacking.5 7 Apart from symptomatic therapy (antidepressants, anticonvulsants, analgesics), current treatment options include alpha-lipoic acid (300–600 mg per day intravenously, for 2–4 weeks) which is known to improve nerve conduction velocity and alleviate positive neuropathic symptoms in patients with DPN;8 this effect is supposedly attributed to its antioxidant action.9 Several other antioxidants, including succinic acid, riboflavin, and nicotinamide, demonstrate positive impact on the nervous system, including reversal of neurological deficit and alleviation of oxidative stress, in experimental diabetes mellitus in animal studies.10–13
A combined metabolic medication, containing succinic acid, inosine, nicotinamide, and riboflavin (SINR; manufactured as Cytoflavin, by ‘POLYSAN’, Russia) promotes glucose utilization and possesses an antioxidant effect. Its administration in experimental diabetes in rats was accompanied by a decrease in the level of circulating products of lipid peroxidation and low-density lipoprotein cholesterol.14 Derivatives of 3-hydroxypyridine and succinic acid were not inferior to alpha-lipoic acid in experimental therapy of behavioral disorders and conditioned reflex learning in alloxan diabetes in rats; in addition, the use of succinic acid in the acute period of alloxan diabetes reduced hyperglycemia and normalized triglyceride levels.15 16 In humans with DPN and diabetic foot syndrome, succinate-containing medications may contribute to reducing the severity of DPN, improving tissue oxygenation, and restoring the activity of enzymes of the antioxidant system.17 18 Antioxidant effect of the combined medication, by analogy with alpha-lipoic acid efficacy, substantiates the hypothesis of its effectiveness in the combined treatment of patients with DPN.
We aimed to evaluate the efficacy and safety of combined metabolic medication, containing inosine, nicotinamide, riboflavin, and succinic acid, compared with placebo in the treatment of patients with DPN.
Materials and methods
Study overview
We performed a randomized double-blind, placebo-controlled clinical trial in 10 clinical centers in Russian Federation.
Study population
We included men and women aged 45–74 years, with diagnosis of type 2 diabetes confirmed ≥1 year before recruitment and previously diagnosed symptomatic distal sensorimotor DPN, as demonstrated by Total Symptom Score19 (TSS) ˃5 points, with a score ≥2 by at least one of the TSS subscales; the severity of pain on the TSS subscale had to be ≤2; and NIS-LL (Neuropathy Impairment Score of the Lower Limb score) ≥2. Diagnosis of DPN was previously established in the ambulatory centers of diabetes control; diagnostic evaluation was not repeated for the study purpose. Other inclusion criteria were body mass index 22–40 kg/m2; hemoglobin A1c (HbA1c) 7.0%–10.0%; regular treatment with oral hypoglycemic drugs and/or insulin of medium, long or ultra-long duration of action and/or agonists of glucagon-like peptide-1 receptors, in stable dosage for at least 12 weeks before screening; consent to maintain a stable diet, exercise, therapy, and diabetes control, and adequate contraceptive methods if fertile. We excluded pregnant or lactating women; patients with type 1 diabetes and other specific types of diabetes; with fasting plasma glucose (FPG) >15 mmol/L; history of ketoacidosis or hyperosmolarity (coma) within 6 months prior to screening, or hypoglycemia within 3 months before screening; severe manifestations of uncontrolled diabetes, including proliferative retinopathy, diabetic foot with limb ischemia or wound defect, neuro-osteoarthropathy; patients on therapy with short-acting and ultra-short-acting insulins, prior or current treatment with systemic corticosteroid drugs, cytostatics, or penicillamine; patients with severe central nervous system disorders (seizures, head trauma, demyelinating disease, tumor), malignancies, decompensated cardiovascular disease currently or within 3 months before screening, kidney disease with a glomerular filtration rate <30 mL/min; active viral hepatitis or cirrhotic liver disease; anemia or acute blood loss or blood transfusion within the previous 12 weeks; HIV infection; any severe infection within 30 days of screening; and history of drug or alcohol abuse. We did not include patients with type 1 diabetes and patients treated with short-acting insulins in order to avoid imbalance and wide variations of insulin daily doses in the study population.
Study medication SINR is a combined product available in two forms: 10 mL vials with solution for intravenous infusion, containing (per 1 L): succinic acid 100 g, nicotinamide 10 g, inosine 20 g, riboflavin sodium phosphate 2 g; and enteric-coated tablets, containing succinic acid 0.3 g, inosine 0.05 g, nicotinamide 0.025 g, and riboflavin sodium phosphate 0.005 g. Intake of inosine, nicotinamide, riboflavin, succinic acid, alpha-lipoic acid, thiamine derivatives, pyridoxine, cyanocobalamin (excluding multivitamins), antidepressants and gabapentin derivatives within 3 months before screening was prohibited during the whole study period. In addition, other drugs with action on the nervous system and metabolic medications were not allowed for use within the whole study period, including antiepileptic drugs, antidepressants, agents affecting neuromuscular transmission, anticholinesterase agents, drugs for the treatment of dementia, psychostimulants and nootropic drugs, centrally acting sympathomimetics and parasympathomimetics, peripheral vasodilators, and drugs containing vitamin B.
Study design
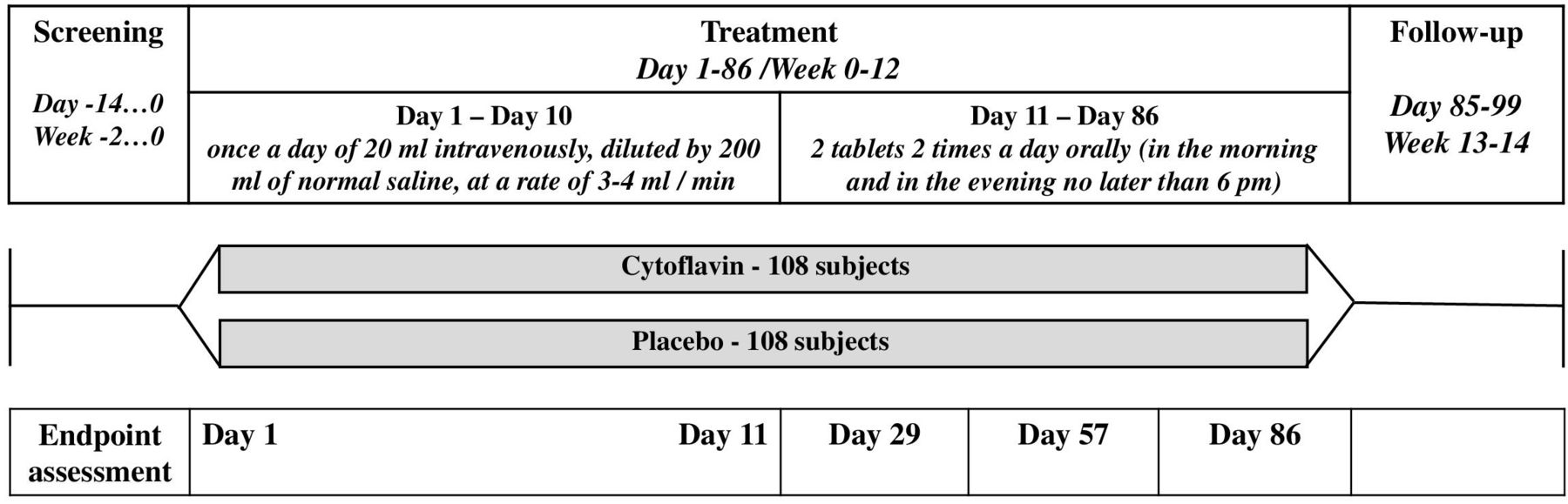
Participants were randomly assigned (1:1) into an experimental group or placebo group using the interactive web response system. Block randomization was used (block sizes of four) stratified by intake of insulin. The randomization sequence was drawn up by an independent, blinded statistician who used a validated pseudo-random number generator. The study consisted of a screening period (2 weeks), a treatment period (12 weeks), and a follow-up period (2 weeks) (figure 1). Treatment was performed in two phases: intravenous infusion of study medication/placebo for 10 days, followed by oral administration of study medication/placebo for 75 days; in total the duration of investigational treatment was 12 weeks or 85 days. During the first phase (days 1–10), the study medication/placebo was administered once a day at a dose of 20 mL intravenously, diluted by 200 mL of normal saline, at a rate of 3–4 mL/min. The solution for infusion was administered in each center by a blinded team, except for the one unblinded person who prepared the medication before infusion; the yellow color of the solution was masked by opaque infusion systems and dark coverings for vials. From day 11, patients were given study medication/placebo for self-administration, to be taken two tablets two times per day orally (in the morning and in the evening no later than 18:00). Blinding of tablets was ensured by masking with a placebo, packaging, and the same labeling. All outcome assessments were performed by the investigators who were blinded to treatment allocation.

A schematic illustration of the study procedures.
Study procedures
The following tests and scales were used for study assessments: general scale for the assessment of neurological symptoms (TSS);19 20 objective assessment of neurological symptoms with NIS-LL;21 rough neuropsychological assessment of executive ability by performing a simple test of visual scanning and working memory, Trail Making Test (TMT);22 evaluation of symptoms of chronic asthenia by Fatigue Assessment Scale (FAS).23 All assessments were performed on days 0, 11, 29, 57, and 86. Details of TSS assessment were published elsewhere.19 20 The primary outcome was improvement of DPN symptoms, as measured by change in TSS at week 12 (day 86) from baseline. Secondary outcomes, also assessed at day 86 from baseline, were changes of the individual components on TSS scale, the proportion of patients who achieved ≥50% reduction in TSS total score, change in NIS-LL score, change of the results of the TMT (part A and part B), change in FAS scores, HbA1c and FPG levels, and changes of the atherogenic index and body weight. Safety outcomes were the incidence of hypoglycemia episodes during the 12 weeks of study treatment, and the incidence of adverse events (AEs) and serious AEs (SAEs) based on subjective complaints, laboratory tests, results of physical examination, vital signs, and ECG. Registration of AEs/SAEs was provided from the signing of informed consent until the completion of participation in the study. Cases of AE were coded using the latest MedDRA medical dictionary (V.23.1). Deviations of laboratory findings from normal ranges, and clinically significant deviations in vital signs and ECG records during the study period were reported as AEs.
Statistical analysis
In a double-blind, randomized, placebo-controlled trial SYDNEY 224 alpha-lipoic acid showed superiority over placebo in patients with DPN in terms of a decrease in the total TSS score after 5 weeks of treatment. The minimum decrease in TSS in patients receiving the study drug was 4.5 points, and in the placebo group 2.9 points. The maximum SD of the TSS change was 3.5 points. For this study, the sample size calculation was based on assumption that the difference between SINR and placebo groups in terms of TSS reduction at week 12 from baseline will be 1.5 points or more. To achieve 80% statistical power with α=0.05 and SD 3.5 points, to test the superiority hypothesis, 86 patients in each group must be included in the analysis. Assuming a 20% potential dropout rate, the calculated study population was 216 patients (108 patients in each group). The primary population for efficacy analysis was the full analysis set (ITT), which included all randomized patients who received at least one dose of study medication and had at least one efficacy parameter score after baseline (intent-to-treat analysis, ITT). In addition, efficacy analysis was performed in the per-protocol (PP) population, which embodies patients who received the full course of investigational treatment without significant protocol deviations. All patients who received at least one dose of the study medication were included into the safety analysis.
Data analysis
A t-test was used to assess the primary efficacy endpoint, that is, the change in the total TSS score at week 12 from baseline. Additionally, analysis of covariance was performed using treatment group, stratum, and center factors as fixed parameters to test the null hypothesis of absence of difference between groups. To assess the dynamics of TSS subscores between visits, a mixed model was used, with the subscale score as a dependent variable, group as a fixed factor, center and subject as random factors, and visit number as a repeated factor. The analysis of other numerical parameters of efficiency was carried out in a similar way. For the analysis of secondary efficacy endpoints, which were categorical variables, and for between-group comparison of safety (AE/SAE), the χ2 test or, where appropriate, Fisher’s exact test, was used. A mixed repeated measures model was used to assess the dynamics of study variables between visits. To assess the dynamics of laboratory parameters in relation to the baseline, a paired t-test or Wilcoxon test (depending on the type of data distribution) was used separately for each group. For between-group comparison of changes relative to baseline, the t-test or the Mann-Whitney test was used, depending on the type of data distribution. Testing of quantitative variables for normality of distribution was carried out using the Shapiro-Wilk test and the test for skewness and kurtosis.
The incidence of AEs/SAEs during treatment was calculated for each class of organ systems and the preferred term for each group. Summary data on the severity of AEs, their association with study medication, and outcomes were presented for each organ system class. Data on SAEs, AEs leading to the need for early completion of the study for the subject, and AEs with severity according to National Cancer Institute Common Terminology Criteria for Adverse Events (V.5.0, publish date: November 27, 2017) of grade 3 or higher were analyzed separately. For between-group comparison of safety parameters, the χ2 test and, where appropriate, Fisher’s exact test, was used. All statistical calculations were performed using Stata V.14.
Results
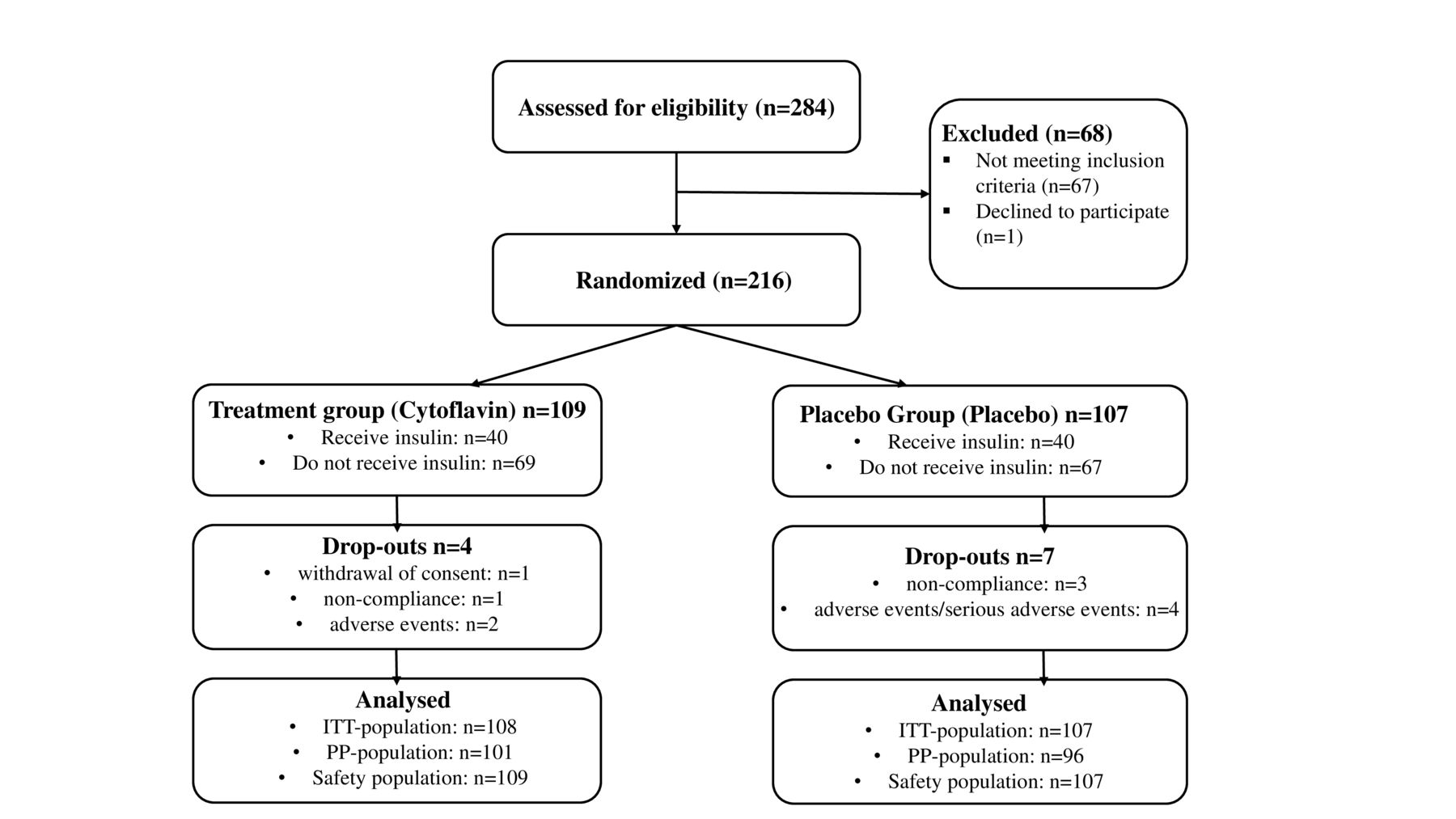
In 2020–2021, 284 patients were screened, of those 216 were randomly allocated to the treatment group (109 of 216; 50.5%), and the placebo group (107 of 216; 49.5%); of those, ITT population comprised 215, PP population 197, and safety population 216 individuals (figure 2). Use of blocks resulted in uneven final distribution of patients (109:107 instead of planned 108:108).
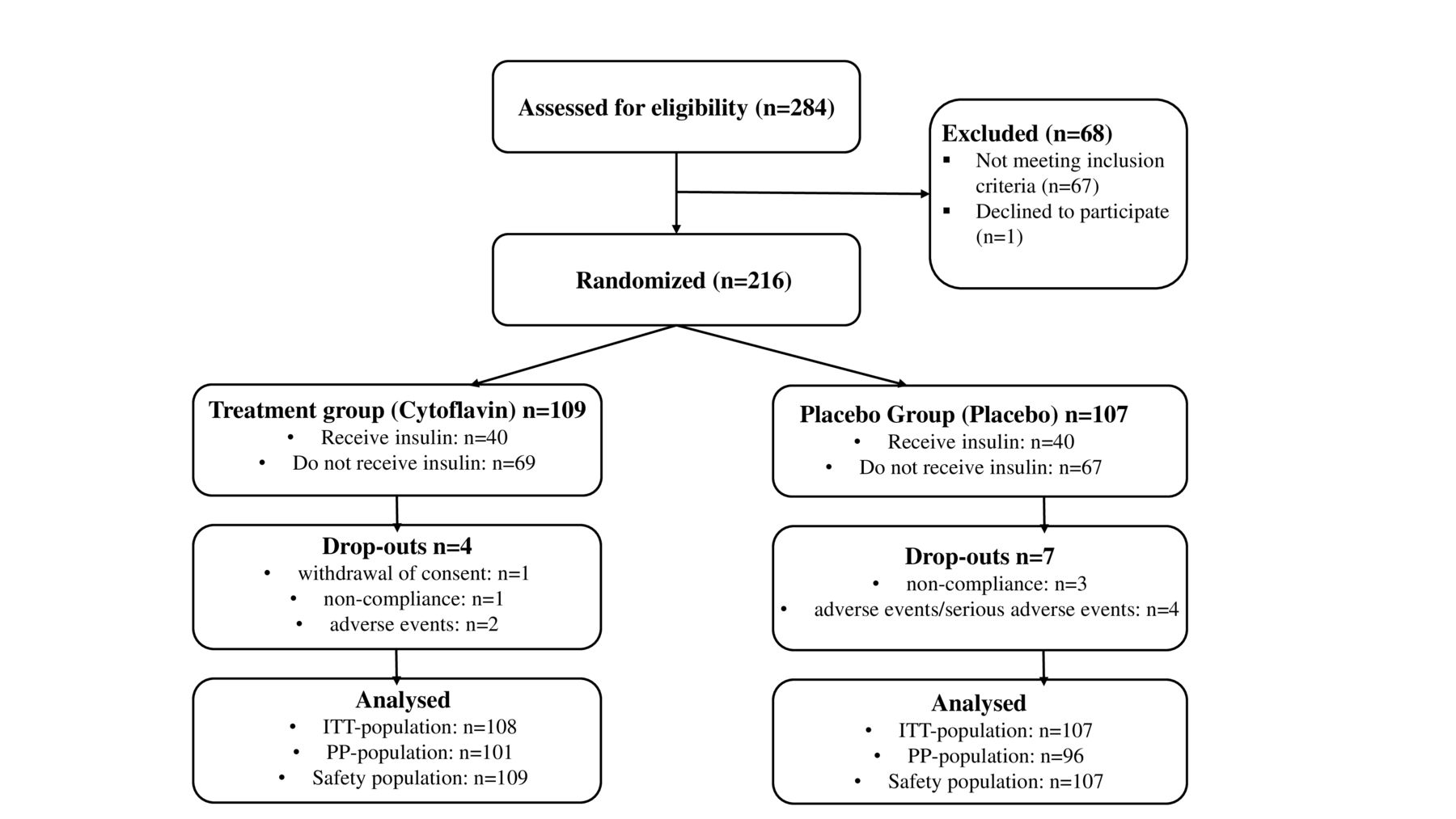
Allocation of study subjects and trial profile. ITT, full analysis set; PP, per protocol.
Description of the study groups
Baseline demographic data, clinical characteristics, and comorbidities assessed in the ITT population were similar between treatment groups (table 1). All study subjects were Caucasians. Statistically significant differences of the incidence of sequelae of diabetes were not observed, except for the presence of diabetic retinopathy, which was more prevalent in the placebo group. Most patients in both groups had type 2 diabetes for more than 10 years. The duration of DPN was, in most cases, longer than 4 years. Only 4 of 108 (3.7%) patients in the SINR group and 3 of 107 (2.8%) patients in the placebo group received concomitant therapy for DPN (p>=0.991). The most frequent comorbidities were arterial hypertension, dyslipidemia, ischemic heart disease, and obesity. All patients received glucose-lowering therapy for type 2 diabetes; also, most patients received antihypertensive therapy and treatment for coronary artery disease. Compliance with the study medication intake in both groups was within the range 80%–120%, as permitted by the study protocol.
Baseline demographic data, clinical characteristics, and comorbidities
Primary endpoint
In the experimental group, a significantly greater reduction in the symptoms of DPN, assessed by the TSS, was achieved, compared with the placebo group (table 2). Reduction of symptoms measured by the TSS scale in subgroups, defined by the degree of compensation for type 2 diabetes (as reflected by glycated hemoglobin level below vs above 8.0%) and by severity of DPN symptoms (as demonstrated by baseline TSS below vs over 7.5), is presented in table 2.
Total Symptom Score (TSS) statistics and the change of the total score by the end of the treatment period, in total study population and in subgroups defined by HbA1c (below 8.0% vs ≥8.0%) and baseline TSS (below 7.5 vs ≥7.5)
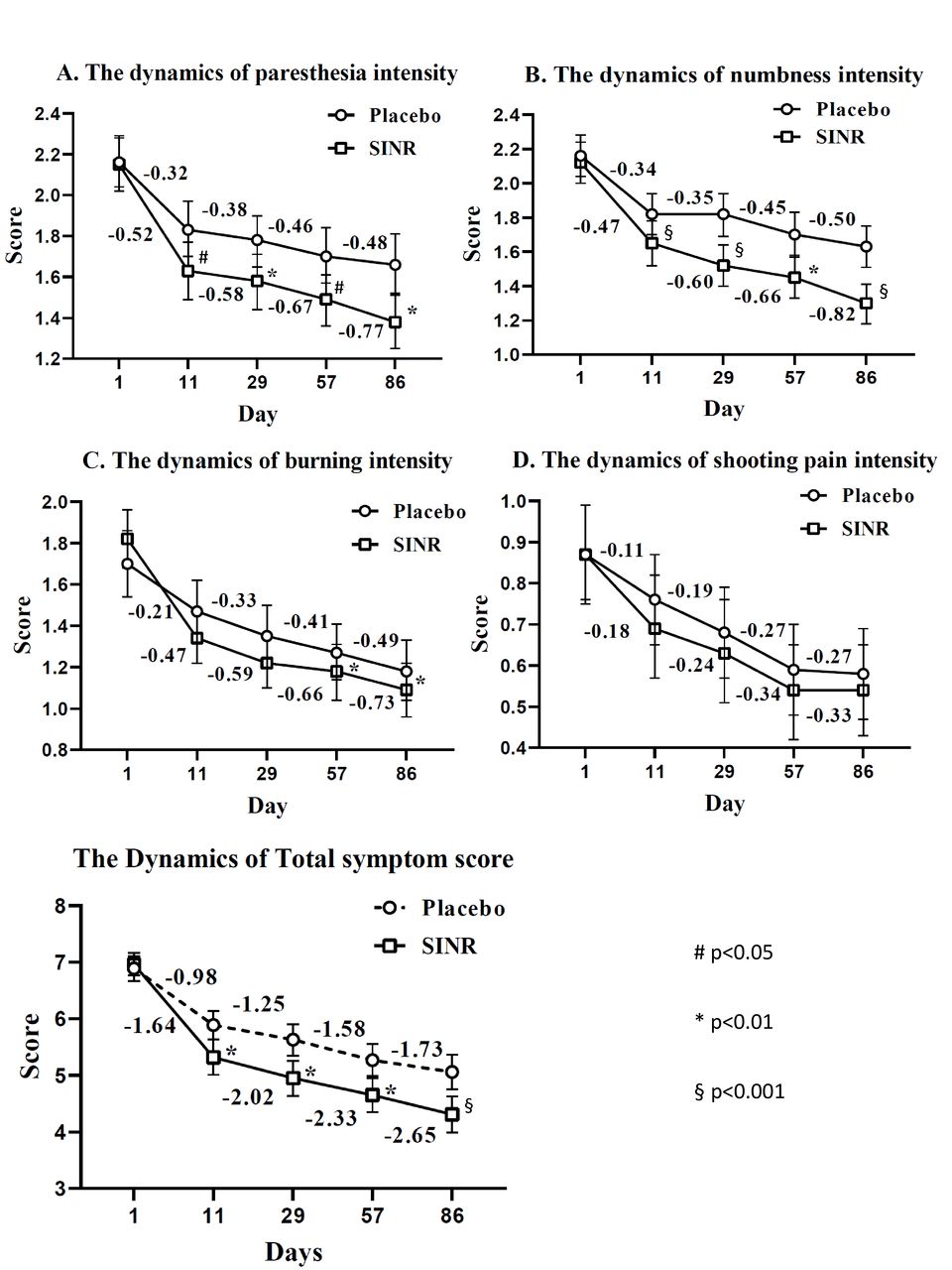
As for the individual components of the TSS, several statistically significant differences were found between groups in terms of reduction of TSS subscores at different visits (figure 3). The analysis revealed the presence of statistically significant differences between the groups by dynamics of the intensity of paresthesia (p=0.004) and the intensity of numbness (p=0.003). There were no statistically significant differences between the groups in the dynamics of the intensity of burning (p=0.245) and the intensity of pain (p=0.578). On day 11, paresthesia intensity was 1.63±0.73 in SINR group and 1.83±0.72 in the control group (p=0.045); on day 31 1.58±0.71 and 1.78±0.66 (p=0.021), on day 57 1.49±0.66 and 1.70±0.69 (p=0.024), and on day 86 1.38±0.67 and 1.66±0.74 (p=0.018), respectively. Intensity of numbness in SINR group versus control group on day 11 was 1.65±0.66 vs 1.82±0.61 (p=0.049), on day 31 1.52±0.62 vs 1.82±0.61 (p=0.006), on day 57 1.45±0.64 vs 1.70±0.65 (p=0.084), and on day 86 1.30±0.60 vs 1.63±0.60 (p=0.022). By the end of treatment, statistically significant differences between the groups were detected also by the intensity of burning: on day 57 1.18±0.67 vs 1.27±0.71 (p=0.001), and on day 86 1.09±0.69 vs 1.18±0.74 (p=0.007) in SINR group versus control group, respectively.
{kind=link}
{kind=link}
{kind=link}
Baseline level and the sequential changes of the Total Symptom Score and its individual components in experimental and placebo groups, based on ITT population. (A) Paresthesia intensity; (B) Numbness intensity; (C) Burning intensity; (D) Shooting pain intensity; (E) Total score (TSS); #p<0.05; *p<0.01; §p<0.001. ITT, full analysis set; SINR, succinic acid+inosine+nicotinamide+riboflavin.
Secondary endpoints
We did not observe statistically significant differences by the other secondary study endpoints. The mean decline of NIS-LL score on day 86, that is, at the end of therapy, compared with baseline in the experimental group was 3.2 points, in the placebo group 2.5 points (p=0.098). Neither statistically significant differences by dynamics of changes in the NIS-LL scale were found between the groups throughout the 12 weeks of treatment (p=0.166, mixed model). However, we observed a non-significant trend of improvement of touch pressure sensation, as reflected by lower mean NIS-LL touch pressure subscore, in patients receiving SINR versus placebo on day 86 (1.32±0.9 vs 1.56±0.8, respectively, p=0.055) (online supplemental table 1). Mean HbA1c levels slightly decreased in both study groups; on day 86 compared with baseline, mean change was −0.36 (95% CI −0.53 to −0.19) in SINR group and −0.23 (95% CI −0.42 to −0.05) in placebo group (p=0.306). In the SINR group, there was also a trend to the decrease in the FPG levels (mmol/L) on day 29, day 57, and day 86 relative to the baseline level, but this trend did not reach statistical significance (p=0.334). The performance indicators of the TMT (execution time, number of errors) of part A and part B improved in both groups, but the analysis did not reveal statistically significant differences between the groups by these parameters. In both treatment groups, there was a slight decrease in the atherogenic index and in body weight, but without statistical significance between the treatment groups (p=0.817 and p=0.755, respectively).
Supplemental material
Safety endpoints
During 12 weeks of investigational treatment, hypoglycemia occurred in 16 of 108 (14.8%) patients of the experimental group and in 25 of 107 (23.4%) patients of the control group (p=0.105). All episodes did not require special treatment (only intake of carbohydrate food or drinks) and resolved without consequences. In addition, during the study, 173 AEs were recorded, which developed in 87 patients; 32 episodes of reported AEs in 16 patients had at least a possible association with investigational treatment. The most common AEs were increased blood pressure (SINR/placebo=11/11), headache (SINR/placebo=19/8), and dizziness (SINR/placebo=6/4). AEs reported by the investigator as related to study drug administration (definite, probable, possible association categories) were hypertension, headache, and dizziness. There were five episodes of SAE, which required hospitalization of patients: four cases of pneumonia (the study period substantially overlapped with the peak of COVID-19 pandemic) and one case of the newly detected atrial fibrillation paroxysm. No fatal AEs were reported. There were no statistically significant differences between the study groups in terms of AE frequencies, severity, outcomes, and measures taken.
Discussion
Oxidative stress is known to be an important factor in the pathogenesis of DPN;25 therefore, the use of medications with antioxidant activity seems to be justified, in particular given that the benefit of intensive glycemic control does not lead to substantial reduce of DPN occurrence in patients with type 2 diabetes mellitus.26 In a randomized, double-blind, placebo-controlled study, we demonstrated the efficacy of the combined medication with antioxidant effect (SINR) against symptoms of DPN. Treatment with SINR, compared with placebo, leads to a significant decrease in the frequency and severity of symptoms of DPN, as demonstrated by statistically significant decrease in the total TSS, both in ITT and in PP populations. The treatment effect was noted early at the beginning of the treatment course (day 11, after the course of intravenous infusions) and sustained over time at each time point (on days 29, 57, and 86). A significant reduction of symptoms in the experimental group was achieved regardless of the degree of compensation for type 2 diabetes, both with the level of glycated hemoglobin below 8% and ≥8%, which corresponds to the previous concept that DPN severity is not directly related to the quality of glycemic control.26 Assessing the treatment effect according to the initial severity of DPN symptoms, it was found that better results of SINR treatment were achieved in patients with milder initial symptoms (TSS <7.5); similar findings were observed in studies of other antioxidant drugs.27
Assessing the results of treatment according to the individual components of the TSS scale (paresthesia, numbness, shooting pain, burning), we found that SINR had a greater effect on reducing paresthesia and numbness in DPN than on the intensity of pain and burning. It should be noted that burning sensation can be defined by the patient as burning pain and is conducted by the same fibers that are responsible for the feeling of acute pain, that is, the thin unmyelinated C-type fibers, unlike paresthesia and numbness, which are mediated when thick myelinated fibers are affected. These results were expected given the mechanism of action and points of application of SINR, which is not a medication for the treatment of neuropathic pain but is an antioxidant substance. However, after 2 months of treatment, its effect was also observed by the ‘burning’ component of TSS scale. While the other medication for pathogenetic treatment of DPN, alpha-lipoic acid, alleviates primarily neuropathic pain,28 SINR may be also considered as an additional treatment option targeted to numbness and paresthesia.
The present study is the first clinical trial of the combination of SINR in type 2 diabetes. The antioxidant potential of this combination was previously demonstrated in other neurological conditions, for example, in intracerebral hemorrhage.29 30 In DPN, clinical effect of SINR was moderate, compared with alpha-lipoic acid; a possible explanation is that alpha-lipoic acid may be a more potent antioxidant, which has an inherent mechanism of direct antioxidation via free radical scavenging and diverse indirect mechanisms,9 including metal chelation, cofactoring mitochondrial antioxidant system, etc. The studied combination of SINR acts mostly in an indirect way: succinic acid inactivates peroxidases in mitochondria and increases the activity of NAD-dependent enzymes, while nicotinamide and riboflavin, as intermediates of electron transport system, enhance the pharmacological activity of succinic acid. The factors studied at the present trial do not provide explanation of the phenomenon of better effect in patients with milder symptoms, but the possible explanation may be lower intensity of lipid peroxidation in patients with less severe DPN. Positive impact of the studied combination on DPN symptoms in the present study, although not very dramatic, may deserve further research, given the lack of treatment alternatives for DPN.
We were unable to demonstrate treatment effect in terms of reduction of objective neurological signs assessed by NIS-LL scale. Of importance, the study was not powered to detect between-group differences in NIS-LL score and its subscores. In our study, experimental treatment reduced the severity of sensory disorders in DPN (assessed by the TSS scale), while having little effect on the ‘negative’ neurological signs, such as mitigation of tendon reflexes at the lower extremities, decrease muscle strength, and a decrease in various types of sensitivity assessed by the NIS-LL scale. It should be noted that, in contrast to the ‘negative’ signs, the symptoms of sensory impairment (paresthesia, numbness, burning sensation, pain) are precisely those DPN manifestations that primarily bother the patient and have a significant impact on the quality of life.
In the present study, treatment with SINR did not increase the risk of hypoglycemic reactions, both during intravenous infusions and oral intake of the tablets; thus, it may be concluded that SINR does not affect the safety of glucose-lowering therapy in patients with type 2 diabetes mellitus, at least those regimens which were not prohibited by the study protocol. Based on the obtained safety data (no significant differences between the experimental group and the placebo group in the number, severity, outcomes of AEs and SAEs, and in frequency of AEs which have at least possible relation to the study treatment), we conclude that SINR (solution for intravenous administration and enteric-coated tablets) has a favorable safety profile.
The limitations of the present study include comparatively small effect size, moderate proportion of patients with severe DPN symptoms, subjective assessment of outcomes, exclusion of participants who received injectable glucose-lowering medications other than medium, long or ultra-long duration of action insulins, as well as patients with uncontrolled diabetes and type 1 diabetes. Recruitment of patients 45–74 years old restrains generalizability of results to other affected age groups. However, a metabolic antioxidant drug with a favorable safety profile, which has demonstrated the ability to alleviate DPN symptoms, may become an additional alternative to DPN therapy, in view of the limited treatment options for this condition.
Conclusion
The combined medication SINR (Cytoflavin) has demonstrated efficacy, as reflected by alleviation of DPN symptoms, and safety in patients with type 2 diabetes mellitus. Taking into consideration the lack of treatment alternatives, a course of intravenous infusions followed by intake of enteric-coated tablets may be recommended for the treatment of symptomatic DPN. Further studies are needed to support the clinical efficacy by the results of instrumental investigations.
Data availability statement
Data may be obtained from a third party and are not publicly available. Data were collected and stored by an independent CRO.
Ethics statements
Patient consent for publication
Ethics approval
The study was conducted in accordance with the requirements of the Guideline for Good Clinical Practice of International Conference on Harmonization (ICH GCP) and the Eurasian Economic Union (EAEU), the principles specified in the Declaration of Helsinki, the current laws of the Russian Federation, and the Protocol of the clinical study CTF-III-DM-2019 (approved by the Russian Ministry of Health (no 109) on March 16, 2020). Prior to the initiation of the clinical trial, written approval from the Local Ethics Committees was obtained for all research sites. All patients have provided written informed consent to participate in the study.
Acknowledgments
The authors would like to thank study participants and all collaborators, including all site coordinators and investigators.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TK—conceptualization, methodology, supervision, formal analysis, and writing (original draft). YGS—investigation and writing (review and editing). AFV—investigation and writing (review and editing). NSO—investigation and writing (review and editing). VPP— investigation and writing (review and editing). IAS—conceptualization, methodology, supervision, and writing (review and editing). TK is responsible for the overall content as guarantor.
Funding This work was supported by POLYSAN Scientific & Technological Pharmaceutical Company. POLYSAN Scientific & Technological Pharmaceutical Company provided funding for this research and reviewed the research prior to submission.
Disclaimer The sponsor was involved in study design and in the decision to submit the article for publication; the sponsor was not involved in the collection, analysis, and interpretation of data.
Competing interests TK was an employee of POLYSAN Scientific & Technological Pharmaceutical Company when this research was conducted. IAS was a scientific advisor for this study and was compensated by POLYSAN Scientific & Technological Pharmaceutical Company for his work on this research. YGS, AFV, NSO, and VPP were compensated by POLYSAN Scientific & Technological Pharmaceutical Company for their work on this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.