Article Text

Abstract
Introduction We compared the efficacy and safety of human regular insulin (HRI) versus rapid-acting insulin (RAI) in a type 2 diabetes population already using the V-Go insulin delivery device.
Research design and methods This was a 14-week, multicenter, randomized, open-label, parallel-group, phase IV, non-inferiority study. Patients ≥21years of age, with inadequately controlled type 2 diabetes who were currently using the V-Go insulin delivery system with RAI, with glycated hemoglobin (HbA1c) ≥6.5% (≥48 mmol/L) to ≤12.5% (≤108 mmol/L) were randomized 1:1 to RAI continuation or switch to HRI. The primary outcome was estimated treatment difference (ETD) in HbA1c least-squares mean change from baseline at 14 weeks (prespecified non-inferiority hypothesis with 95% CI upper limit <0.4%). Primary analysis was by per protocol (PP); safety analysis was by intention to treat.
Results We randomized 136 patients to continued RAI treatment (n=67) or HRI (n=69); 113 patients were included in the PP analysis (RAI, n=54; HRI, n=59). Mean change in HbA1c from baseline to study end was −0.60±1.1% (95% CI −0.90 to –0.29); −6.6±12.0 mmol/mol (95% CI −9.8 to −3.2) with HRI treatment and −0.38±1.3% (95% CI −0.70 to –0.05); −4.2±14.2 mmol/mol (95% CI −7.7 to −0.5) with RAI treatment, with ETD of −0.22% (95% CI −0.67 to 0.22); −2.4 mmol/mol (95% CI −7.3 to 2.4), p=0.007, confirming non-inferiority of HRI to RAI. No between-group differences in changes in total daily insulin doses, number of hypoglycemic values (≤70 mg/dL (≤39 mmol/L) or body weight were observed. No severe hypoglycemic events were reported. Direct pharmacy cost savings (−US$265.85; 95% CI −US$288.60 to −US$243.11; p<0.0001) were observed with HRI treatment.
Conclusions Individuals with type 2 diabetes requiring insulin can be treated with V-Go wearable insulin delivery device using HRI, safely and effectively, and potentially at a much lower cost compared with RAI, which can lead to improved access to insulin therapy for these individuals.
Trial registration number NCT03495908.
- diabetes mellitus
- type 2
- insulin
- insulin infusion systems
- costs and cost analysis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Recent studies using continuous subcutaneous insulin infusion (CSII) have demonstrated significant glycemic and quality of life benefits compared with multiple daily insulin injection therapy in type 2 diabetes; however, current CSII devices are only approved for use with increasingly expensive rapid-acting insulin (RAI) analogues.
What are the new findings?
Findings from the current non-inferiority study suggest that use of human regular insulin (HRI) administered via the V-Go device is a clinically viable and more cost-effective option than using RAI, which could lead to more robust savings as compared with RAI.
How might these results change the focus of research or clinical practice?
Our findings may prompt clinicians to consider use of HRI via delivery methods such as the V-Go device in patients with insulin-treated type 2 diabetes.
Introduction
Insulin has the advantage of being effective where other agents are not and is commonly used in patients with type 2 diabetes for the management of persistent hyperglycemia. Use of basal-bolus insulin therapy in patients with insulin-treated type 2 diabetes provides a more physiological approach than basal insulin alone to improve glycemic control.1 2 Although intensive insulin management can help slow the progression or even prevent long-term complications associated with poor glycemic control, treatment adherence among patients who initiate basal-bolus therapy is often suboptimal.
Several factors contribute to treatment non-adherence,3 the affordability of insulin, particularly analogue insulins, has become a major challenge for many patients with diabetes in the USA.4 Since 2001, the cost of a 10 mL vial of insulin has increased almost 588%.5 As a result, individuals are often forced to choose between purchasing their insulin and daily necessities.6 7 Many individuals are now rationing their insulin, resulting in poor glycemic control with an increased risk of severe complications.8
Numerous studies comparing rapid acting insulin (RAI) with regular human insulin (HRI) treatment in type 2 diabetes populations have shown similar reductions in glycated hemoglobin (HbA1c) in both study groups with no differences in long-term outcomes, severe hypoglycemia or other adverse events (AEs).9–14 Yet, the price of RAI formulations is significantly higher than HRI which can have negative implications for health plans and patients. When using published wholesale acquisition to normalize costs across insulin types, HRI formulations range from US$138 to US$149 per vial compared with US$275 to US$289 for RAI formulations, before any health plan discounts or rebates are applied. Patients paying cash for insulin may encounter a range from US$28 to US$99 per vial for HRI formulations, whereas prices for RAI formulations range from US$178 to US$562 per vial based on a nationally recognized website that compares pharmacy pricing and discounts.15 Given the high cost of RAI compared with HRI and lack of data demonstrating clinically significant differences between the formulations, the use of HRI, particularly when administered with a modern insulin delivery device, is a viable clinical option in managing insulin-treated type 2 diabetes and may help reduce the cost burden of diabetes management for many health plans and make insulin more affordable for patients without sacrificing glycemic control.
However, affordability of insulin is only one factor impacting patient willingness to initiate and adhere to intensive insulin therapy. Many patients are reluctant to add prandial insulin due to fear of hypoglycemia,16 17 weight gain,17 18 treatment complexity3 16 17 19 and potential for embarrassment when administering insulin in public places.20 21
Use of insulin pumps has the potential to address many of these issues and improve glycemic control in patients with type 2 diabetes.22 However, because current insulin pumps include complex features that patients with type 2 diabetes may find unnecessary and difficult to use, some manufacturers have developed disposable patch-like insulin delivery devices that offer the advantages of simple and discreet delivery of both basal and/or bolus components at a lower cost than conventional insulin pumps.
One example of this technology is the V-Go wearable insulin delivery device (Zealand Pharma, Søborg Denmark), a fully mechanical, disposable patch-like device that delivers a continuous preset basal rate of either 20, 30 or 40 units (U) in one 24-hour period, as well as on-demand bolus of up to 36 U in 2 U increments.23 After the device is filled with insulin and affixed to the skin, the user pushes a button to insert a 4.6 mm, 30-gauge stainless steel needle subcutaneously, which initiates delivery of a basal rate of insulin. On-demand prandial insulin doses can be administered at meals by pressing the bolus-ready button and the bolus-delivery button through clothing, allowing for discrete insulin administration. The device is removed and replaced with a new device every 24 hours.
Several studies have demonstrated the V-Go device is a safe and effective option for insulin delivery in patients with suboptimally controlled type 2 diabetes.21 24–28 These studies showed significant improvement in glycemic control,21 24–28 with lower insulin requirements,24 27 and lower cost24 27 compared with multiple daily insulin injection (MDI) with similar hypoglycemic events.21 24–28 Although the V-Go device is currently indicated for use with U-100 RAI analogues, studies have shown that U-100 HRI is stable in V-Go devices29 with demonstrated efficacy and safety in a small retrospective evaluation.30
The aim of this study was to assess the clinical effects, safety and associated insulin costs of using U-100 HRI compared with a RAI analogue in patients with type 2 diabetes who are currently using the V-Go device for basal-bolus therapy.
Research design and methods
Study design
This was a pragmatic, randomized, open-label, parallel-group, multisite, phase IV, non-inferiority study, comparing the efficacy and safety of using with U-100 HRI compared with U-100 RAI in a population with type 2 diabetes already using the V-Go insulin delivery device. Data points were collected from study participants between April 9, 2018 and August 12, 2019. Participants were recruited from multiple specialized diabetes practices in the USA. All sites had experience in conducting clinical trials.
Participants
Inclusion criteria were: age ≥21 years; ≥6 months since diagnosis of type 2 diabetes; HbA1c ≥6.5% to ≤12.5% (48 mmol/L to 108 mmol/L) at screening; stable dosage of U-100 RAI (<20% change in the 30 days prior to randomization); completed a 7-point glucose profile prior to randomization visit; willing to complete two additional 7-point glucose profiles during the study and able to cover the initial investment and ongoing cost of the insulin delivery device, insulin (RAI or HRI), personal glucometer and supplies for the length of the study.
Exclusion criteria were: confirmed type 1 diabetes; >1 episode of severe hypoglycemia within the 3 months prior to screening; history of hypoglycemia unawareness; pregnant, planning to become pregnant or lactating; use of any oral, injectable or intravenous steroids within 8 weeks prior to screening visit, or plans to take these medications during the study duration; use of U-100 HRI or U-500 HRI delivered by study device within 90 days of screening or medical or other issues that would render study participation unsafe.
Randomization and masking
Following enrollment, eligible patients were randomly assigned in a 1:1 ratio to continue their current RAI insulin (RAI group) or switch to HRI (HRI group), using a computer-generated blocked randomization scheme. Randomization was stratified by study site and baseline HbA1c (<9.0% (75 mmol/mol) and ≥9.0% (≥75 mmol/mol)). Both study groups continued use of the study device for insulin delivery. An open-label design was used and participants, study physicians, investigators and clinical staff were not masked to treatment allocation. The study statistician and sponsor personnel were masked to the treatment allocation until after database lock and analyses were completed.
Procedures
The 14-week trial involved four study visits: screening (V1, week −2); baseline (V2, day 0) and two therapy visits (V3, week 2; V4, week 12). At the screening visit (V1), written informed consent was obtained and medical histories, concomitant medications, HbA1c level, vital signs and physical measurements were documented. Females who were of childbearing potential were given a pregnancy test. Patients were provided a logbook and instructed to use their current glucose meter to perform self-monitoring of blood glucose (SMBG) and document blood glucose values, measuring a minimum of two blood glucose readings daily (fasting and either a pre-evening meal or prebedtime). Participants were also instructed to perform and document 7-point SMBG profiles (preprandial and 2-hour postprandial at all main meals and prebedtime) within 3 days of the baseline visit (V2). Subjects were encouraged to check blood glucose levels if they experienced symptoms of hypoglycemia.
At the baseline visit (V2), eligibility was reconfirmed, and patients were randomized. Logbooks were reviewed, glucose meters downloaded and medical histories, concomitant medications, vital signs and physical measurements were documented. Patients were reminded to perform SMBG and document blood glucose values for the remainder of the study and perform and document 7-point SMBG profiles within 3 days prior to each subsequent therapy visit (V3, V4). Patients randomized to HRI therapy were instructed to administer their prandial insulin doses 20 min before meals and snacks. Patients in the RAI group were instructed to dose 5 min before meals and snacks. Throughout the study, patients were instructed to document the time of the meals, their daily insulin doses and the timing of the insulin dose in their logbooks.
At each therapy visit, investigators reviewed overall glucose control and insulin doses were to be adjusted when deemed appropriate to be consistent with good medical practice and to target glycemic control goals: fasting/preprandial glucose, 80 to <130 mg/dL (4.4 to <7.2 mmol/L); peak postprandial glucose,<180 mg/dL (<10.0 mmol/L)). Glucose meters were downloaded and logbook data (daily SMBG, 7-point profiles, insulin doses, hypoglycemia) were assessed. If consistent or repeated blood glucose levels were lower than the target range and/or frequent episodes of postprandial or fasting/preprandial hypoglycemia were recorded, the patient’s physical activity and carbohydrate intake were to be assessed prior to considering a reduction in prandial insulin dose. Prandial hypoglycemia was to be addressed by lowering the bolus doses and adjusting the basal rate based on fasting glucose and occurrence of nocturnal hypoglycemia or severe hyperglycemia. All hypoglycemia episodes recorded in the patient logbooks or obtained from the patient’s glucometer were logged on the patient source documents and included in the database for evaluation.
At study end (V4), investigators documented concomitant medications, physical measurements, HbA1c levels and vital signs. Logbook data were assessed, and insulin doses adjusted as needed. A central laboratory (Quest Diagnostics) was used to analyse HbA1c levels at V1 and V4 to provide a single standardized testing facility between participating study sites. AEs were assessed and documented at all study visits.
Outcomes
The primary efficacy outcome for testing non-inferiority of HRI was the between-group difference in HbA1c change after 12 weeks of treatment within the per-protocol (PP) population defined as the population who continued the assigned intervention as randomized for the duration of the study period, completing the end of study week 14 visit (visit 4). Among secondary measures, between-group differences in direct pharmacy insulin costs were evaluated at study end in the PP population. All insulin costs are normalized by calculating a 30-day insulin requirement based on total prescribed insulin daily dose and multiplying the monthly insulin dose in units by the unit costs of the prescribed insulin. Costs of insulin are based on current published wholesale acquisition costs and reported in US dollars. Other secondary measures were assessed in the intent-to-treat (ITT) population, which included all randomized participants who received at least one dose of a study medication. These measures included changes and between-group differences in total daily insulin doses (units/kg and units/day) and self-reported hypoglycemia events based on 7-point glucose blood values defined by the International Study Group as: level 1, a glucose alert value of ≤70 mg/dL; level 2, a glucose alert value of <54 mg/dL and level 3, denotes severe cognitive impairment requiring external assistance for recovery.
Safety measures were assessed in the ITT population. These measures included reported hypoglycemia as defined above from both daily and 7-point glucose profiles. AEs were reported as mild, moderate and serious AEs, categorized as not related, possibly related, probably related and definitely related to the intervention.
Statistical analysis
As prespecified in the study protocol, the primary analysis comparing HbA1c response was performed on the PP cohort to avoid bias towards the null hypothesis, which can occur when testing non-inferiority hypothesis in ITT analysis. The non-inferiority of the HRI treatment strategy compared with RAI regimen was assessed using a 95% CI for the between-treatment group net difference (visit 4 minus visit 1) in HbA1c at end of study. This 95% CI was derived from the differences of least square means estimated from a mixed-effects model repeated measures analysis. The study patient was modeled as a random effect. Non-inferiority of HRI treatment was concluded if the upper limit of the 95% CI was less than the non-inferiority margin of 0.4% which is well established in the diabetes field and is supported by the FDA’s guidance on non-inferiority trials in diabetes.31 The sample size calculation assumed an SD of 0.9% for HbA1c at 12 weeks of treatment, using a two-sided significance level of 0.05, and 80% power. Based on our experience with similar populations and length of study, we determined that randomization of 180 patients was required, assuming a 10% drop-out rate.
A planned interim analysis PP was conducted when 75% of the trial participants (n=136) were randomized, using PASS 15 Power Analysis and Sample Size Software 2017 (NCSS, Kaysville, Utah, USA, ncss.com/software/pass). The Lan-DeMets alpha spending function analogue of O’Brien-Fleming group sequential boundaries were used as guidelines for early study termination. A Data and Safety Monitoring Committee (DSMC) reviewed unblinded efficacy and safety data generated for this analysis and reported to the investigators that the data analyzed demonstrated non-inferiority with respect to change in HbA1c over the 12 weeks of treatment, therefore recommending stopping further participant screening and randomization. Additionally, because no untoward safety signals were seen, the DSMC was supportive of early termination of the active randomized participants (n=9) if decided on by the investigators. Based on the interim analysis, the investigators decided unanimously to undergo early termination procedures and the study was terminated.
The secondary efficacy outcome and the safety analyses were performed on the ITT population of 136 subjects. Secondary analyses assessed baseline covariates and their association with treatment efficacy. Group comparisons and changes from baseline over time (study visits) of continuous secondary outcome variables were analyzed with mixed-effects model repeated measures analysis. Hypoglycemic events were summarized as event rate/person week and percentage of patients with at least one event; between-group differences were compared with Poisson repeated measures models using generalized estimating equations and Fisher’s exact test, respectively. Poisson models were selected for hypoglycemic event analysis following tests for overdispersion and assessment of negative binomial models. Safety end points and other AEs were summarized in detail with descriptive statistics. The analysis of safety data was performed for the ITT population. For the primary outcome, the two-sided boundary p value for the interim analysis was 0.015. Secondary analyses were all prespecified and are not adjusted for multiplicity. Statistical analyses were performed with SAS software V.9.4 (SAS Institute, Cary, North Carolina, USA), particularly Proc Mixed for linear models with both fixed and random effects.
Results
Two hundred and seventeen patients were assessed for eligibility, 136 were randomized and 113 completed PP. Among the 81 patients not randomized, the leading reasons were unwillingness to complete 7-point profiles, HbA1c not in study range and unwillingness to participate in the study. A total of 23 patients either withdrew or underwent early termination of the study. Early study termination of nine active study participants was based on a decision reached by the investigators following conclusions from the DSMC that no untoward safety signals were observed from the planned interim analysis. Disposition of study patients is presented in figure 1. The full primary analysis dataset for non-inferiority consists of data from 113 participants (HRI group, n=59; RAI group, n=54) who completed the baseline and therapy visits according to protocol. Baseline characteristics for the PP population were similar between cohorts (table 1) and the characteristics for the ITT population are shown in online supplemental table 1.
Supplemental material
Demographic characteristics of per-protocol population

Disposition of study participants. HbA1c, glycated hemoglobin; HRI, human regular insulin; ITT, intent-to-treat; PP, per protocol; RAI, rapid acting insulin; SMBG, self-monitoring of blood glucose.
Primary outcome
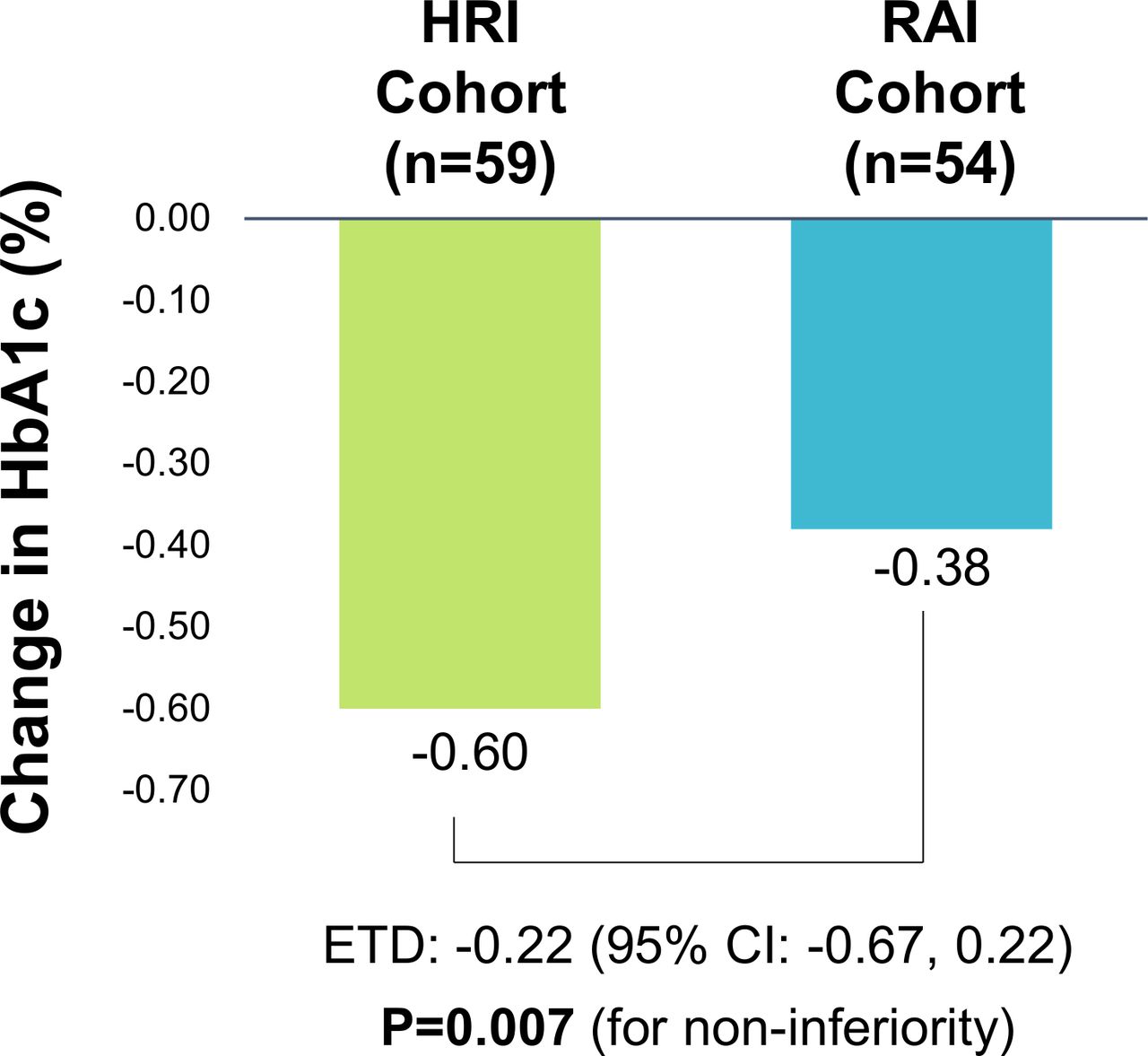
HbA1c significantly decreased from baseline in both treatment cohorts within the PP population. The primary analysis for evidence of non-inferiority of HRI, conducted in the PP population evaluated the upper bound of the two-sided 95% CI for the between-group difference in change in HbA1c compared with the prespecified non-inferiority margin. The estimated treatment difference (ETD) between the two groups was −0.22% (95% CI −0.67 to 0.22) p=0.007 for non-inferiority (figure 2). Based on the 0.015 significance level of the interim analysis, the 98.5% CI for the ETD was (−0.78 to 0.33).

Changes in HbA1c in the PP population. Changes and ETD in HbA1c from baseline. ETD are mean change (95% CI) derived from a mixed model analysis. Upper limit of 95% CI is <non-inferiority margin of 0.4%. ETD, estimated treatment difference; HbA1c, glycated hemoglobin; HRI, human regular insulin; PP, per protocol; RAI, rapid acting insulin.
Secondary outcomes
The secondary efficacy analysis in the ITT population further supported non-inferiority of HRI with an ETD −0.13 (95% CI −0.58 to 0.32), p=0.02. Between-group differences in changes in total daily insulin doses were not statistically different, and post hoc analysis showed no between-group differences in changes in body weight. (table 2).
Changes in total daily insulin dose (U/day, U/kg) and body weight for ITT population
Analysis of the 7-point profiles within the ITT population (n=136) showed no significant differences in the change from baseline in the number of participants with documented hypoglycemia (≤70 mg/dL (≤3.9 mmol/L)) between the HRI (n=2 (2.90%) and RAI (n=4 (5.97%) groups at study end, p=0.44. Event rates (per person week) for level 1 (≤70 mg/dL or (<3.9 mmol/L)) and level 2 hypoglycemia (<54 mg/dL (<3.0 mmol/L)) decreased in both treatment groups when comparing prerandomization to postrandomization (table 3).
Hypoglycemia event rate per person week based on 7-point profiles defined by level for ITT population
The HRI/ RAI between-group hypoglycemia incident rate ratio (IRR) was 0.925 (95% CI 0.386 to 2.217); p=0.86. IRR <1.0 favors the HRI group and an IRR >1.0 favors the RAI group.
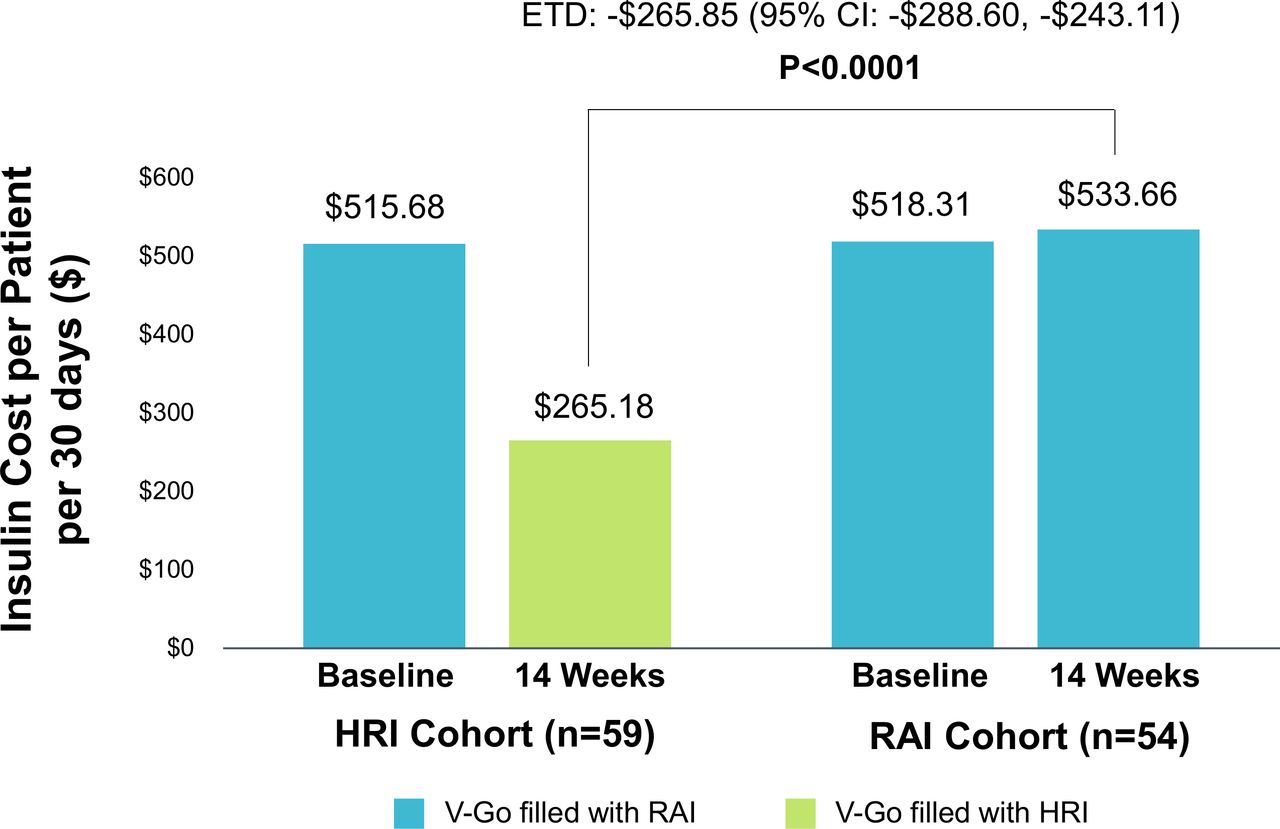
Treatment with HRI resulted in significantly lower insulin costs compared with baseline with a mean change in 30-day insulin costs of −US$250.50 from baseline, p<0.0001 compared with a slight increase in 30-day insulin costs of US$15.35 from baseline in the RAI treatment group, p=0.0668. The ETD between groups for 30-day insulin cost at study end was significantly lower in the HRI treatment group (figure 3).

{kind=link}
{kind=link}
{kind=link}
Pharmacy budget impact in PP population. Baseline and 14-week insulin costs per patient. Insulin cost is based on 30-day costs for HRI and RAI using published wholesale acquisition costs from ProspectoRx (database online). Elsevier, 2020, Tampa, Florida, USA (https://prospectorx.com/Home.aspx). Accessed November 28, 2019. At baseline, RAI was used to fill V-Go by both cohorts. ETD, estimated treatment difference; HRI, human regular insulin; PP, per protocol; RAI, rapid acting insulin.
Safety analysis
Within the safety (ITT) population (HRI, n=69; RAI, n=67), the per cent of subjects reporting hypoglycemia was similar between groups when comparing prerandomization (HRI 33%; RAI 28%) and postrandomization (HRI 41%, RAI 45%) when combining both 7-point profiles and the daily SMBG logs. Event rates (per person week) for level 1 and level 2 hypoglycemia decreased in both treatment groups when comparing prerandomization with postrandomization for all glucose logs. Level 1 hypoglycemia decreased from 0.424 to 0.271 and from 0.332 to 0.187 events/person week for HRI and RAI, respectively. Level 2 hypoglycemia decreased from 0.208 to 0.098 and from 0.049 to 0.041 events/person week for HRI and RAI, respectively. No severe hypoglycemic events or intervention-related moderate or serious AEs were reported in either group. A total of eight patients (HRI, n=5; RAI, n=3) reported a serious AE postrandomization. One mild event (‘upset stomach’) possibly related to the intervention was reported in the HRI group and two mild events (skin irritation and a welt at the needle insertion site) definitely related to the intervention were reported in the RAI group. After a colonic perforation, one patient in the HRI group died following multisystem organ failure which was assessed by investigators to not be related to the study intervention.
Conclusions
This is the first randomized, comparative study of HRI versus RAI efficacy and safety in individuals with type 2 diabetes using a wearable patch-like CSII device. Findings from our study show that delivery of HRI via the V-Go insulin delivery device is non-inferior to RAI in lowering HbA1c; both study groups experienced significant reductions in HbA1c, with similar changes in total daily insulin dose and body weight. Treatment with HRI demonstrated significant cost savings compared with RAI. These findings fully support our study aim to establish the efficacy, safety and cost-efficiency of HRI in the V-Go device.
A key strength of our study was the use of a head-to-head design, which facilitated a direct comparison of the efficacy and safety of the two study insulins to show non-inferiority of HRI compared with RAI therapy. It is important to point out that this effect was observed in patients already using RAI and V-Go device prior to randomization. Another strength was the pragmatic nature of the study, which closely mimicked normal clinical practice for managing type 2 diabetes mellitus. Moreover, our study design and conduct are similar to historical evidence establishing insulin efficacy and delivery and with our results aid in preserving the trial assay sensitivity for concluding non-inferiority. Certain limitations are notable. Most importantly, the open-label nature of the study may have introduced bias to the results. Treatment allocation could not be blinded as the recommended timing of insulin administration varied between HRI and RAI due to differences in the insulin pharmacokinetics. It was not possible to assess persistence with HRI treatment or long-term outcomes due to the short duration of the study. Related to this is the lack of patient-reported outcome data, specifically treatment satisfaction, which is associated with treatment adherence.32 33 While this aspect was not assessed, it is possible that treatment satisfaction may have been lower among patients who switched over from RAI to HRI therapy due to changes in the timing of their prandial dosing; from 5 to 20 min prior to meals and snacks. However, this did not appear to impact glycemic control or participant retention in the HRI study group. Another limitation was allowing patients to use their current blood glucose meters during the study. Variations in accuracy and precision may have impacted the reliability of glucose values obtained and reported. Finally, the pharmacy budget impact may not be generalizable to all health plans or patients prescribed insulin as the cost analysis was based on wholesale acquisition costs due to the lack of drug pricing transparency by manufacturers, the substantial inconsistencies in rebates and discounts granted to health plans and to the wide variations in copays or deductibles paid by patients.
Nevertheless, results from our study are significant given the current costs of RAI therapy and the impact of these costs on medication adherence. In a recent survey of 354 individuals with insulin-treated diabetes, 51 (25.5%) reported underuse of insulin due to cost,8 which aligns with a consistently demonstrated strong link between suboptimal adherence and poor health outcomes and associated costs.3 34–36
Findings from this study demonstrate that use of HRI administered via the V-Go device is a non-inferior, safe, clinically viable and more cost-effective option than RAI treatment in patients with insulin-treated type 2 diabetes. Although these findings suggest that the lower cost HRI may improve treatment adherence, pragmatic studies are needed to assess adherence, persistence and long-term outcomes in real-world clinical settings.
Acknowledgments
The authors would like to thank Christopher G. Parkin, MS, CGParkin Communications, Inc. and Ildiko Lingvay, MD, MPH, MSCS, UT Southwestern Medical Center, Dallas, Texas for their editorial assistance in developing this manuscript and the authors acknowledge the Data Monitoring Committee (DMC) comprising Juan Pablo Frias, MD, FACE, National Research Institute, Los Angeles, California, Rodolfo Galindo, MD, Emory University School of Medicine, Atlanta, Georgia and Song Zhang, PhD, UT Southwestern Medical Center, Dallas, Texas for protecting participant safety and data quality and overseeing the conduct and interpretation of the interim analyses.
References
Footnotes
Presented at Parts of this study were presented at the 13th International Conference on Advanced Technologies and Treatments for Diabetes, Madrid Spain, February 19–22, 2020 and the 80th Scientific Sessions of the American Diabetes Association, Virtual Meeting, June 2020.
Contributors PFM contributed to the design and coordination of the study, conduct of the study, interpretation of the data and the writing of the first and all subsequent manuscript drafts. DRS, AG, BB, JS II contributed to the study design, conduct of the study, data collection and participated in the review and editing of the manuscript. RFG contributed to the study design and coordination of the study, conduct of the study, data collection and participated in the review and editing of the manuscript. CN provided input on the study protocol and contributed to the writing of the first and all subsequent manuscript drafts and preparation of manuscript figures and tables. BA-H contributed to the design of the study, statistical plan and randomization scheme and conducted the analysis, interpreted the data and reviewed and edited the final manuscript. PFM and BA-H had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors approved the final version of the manuscript before submission.
Funding Valeritas, Inc. (acquired by Zealand Pharma) funded this study.
Competing interests PFM reports grants from Valeritas, Inc., and consulting fees from Janssen Pharmaceuticals, Novo Nordisk and Merck. DRS received grants from Valeritas during the conduct of the study; consulting fees from Valeritas, Novo Nordisk, Lilly USA, Boehringer Ingelheim, Janssen, AbbVie, Amarin. AG reports grants from Valeritas during the conduct of the study; consulting fees from Valeritas, Inc., Janssen Pharmaceuticals, Novo Nordisk and Merck. BB reports grants from Valeritas, Inc. acquired by Zealand Pharma during the conduct of the study; consulting fees from AstraZeneca, Boehringer Ingelheim, Novo Nordisk, Valeritas Eli Lilly, Amarin, Senseonics. RFG reports no financial disclosures. CN and JS II are employees of Zealand Pharma. BA-H reports consulting fees from Valeritas, Inc. acquired by Zealand Pharma during the conduct of the study.
Patient consent for publication Not required.
Ethics approval The study was conducted in accordance with Good Clinical Practice (GCP), in accordance with the United States Code of Federal Regulations, Title 21, Part 50 (21CFR50). The clinical study protocol was approved by a central institutional review board (Advarra Institutional Review Board (formally Schulman IRB)) (# 201802193). All participants provided written informed consent prior to enrollment in the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.