Article Text

Abstract
Objective Diabetic nephropathy (DN) is the leading cause of chronic kidney disease and end-stage renal disease. Emerging evidence suggests that complement activation is involved in the pathogenesis of DN. The aim of this study was to investigate the pathogenic role of C3a and C3a receptor (C3aR) in DN.
Research design and methods The expression of C3aR was examined in the renal specimen of patients with DN. Using a C3aR gene knockout mice (C3aR−/−), we evaluated kidney injury in diabetic mice. The mouse gene expression microarray was performed to further explore the pathogenic role of C3aR. Then the underlying mechanism was investigated in vitro with macrophage treated with C3a.
Results Compared with normal controls, the renal expression of C3aR was significantly increased in patients with DN. C3aR−/− diabetic mice developed less severe diabetic renal damage compared with wild-type (WT) diabetic mice, exhibiting significantly lower level of albuminuria and milder renal pathological injury. Microarray profiling uncovered significantly suppressed inflammatory responses and T-cell adaptive immunity in C3aR−/− diabetic mice compared with WT diabetic mice, and this result was further verified by immunohistochemical staining of renal CD4+, CD8+ T cells and macrophage infiltration. In vitro study demonstrated C3a can enhance macrophage-secreted cytokines which could induce inflammatory responses and differentiation of T-cell lineage.
Conclusions C3aR deficiency could attenuate diabetic renal damage through suppressing inflammatory responses and T-cell adaptive immunity, possibly by influencing macrophage-secreted cytokines. Thus, C3aR may be a promising therapeutic target for DN.
- C3a receptor
- diabetic nephropathy
- inflammation
- microarray
- T cell
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
The complement activation is known to be involved in the pathogenesis of diabetic nephropathy (DN).
What are the new findings?
C3aR−/− diabetic mice exhibited less degree of renal damage compared with wild-type (WT) diabetic mice.
By microarray bioinformatic analysis, the inflammatory response and T-cell adaptive immunity were restrained in C3aR−/− diabetic mice compared with WT diabetic mice.
The effects of C3a–C3aR were possibly mediated by influencing macrophage-secreted cytokines.
How might these results change the focus of research or clinical practice?
These findings suggest that C3a might be a bridge linking innate immunity and adaptive immunity in DN, and C3a might be a promising therapeutic target for DN.
Introduction
Diabetic nephropathy (DN) is a prevalent microvascular complication of diabetes mellitus, characterized by microalbuminuria at the early stage that progresses to end-stage renal disease (ESRD) over time. In parallel with the growing incidence of type 2 diabetes, DN remains the most common cause of chronic kidney disease and ESRD.1 2 Cumulating evidence has indicated that inflammatory processes mainly mediated by innate immunity is a key pathophysiological mechanism in the development of DN.3 4 As a major component of innate immunity, the role of the complement system in DN has drawn increasing attention.5
The complement system can be activated through three pathways including classical pathway, lectin pathway and alternative pathway. Activation of all three pathways results in the formation of a C3 convertase which cleaves C3 into C3a and C3b. C3a is a potent anaphylatoxin involved in allergic asthma, allograft rejection, neurodegenerative disease and cancer, signaling through interacting with 7-transmembrane spanning G-protein coupled C3a receptor (C3aR). The C3aR is mainly expressed on cells of myeloid origin including neutrophils, monocytes/macrophages, basophils, eosinophils, mast cells, dendritic cells and microglia,6 as well as non-myeloid cells such as renal tubular epithelial cells.7
A number of studies found an increase of renal C3 expression in patients with DN or diabetic animal models.8 9 Furthermore, in individuals from the general population, high baseline concentration of C3 was related to increased risk of DN.10 However, the role of the downstream fragment C3a in DN is not fully clear yet. In our previous studies, it was found that both plasma and urinary C3a levels were significantly increased in patients with DN, and the urinary levels of C3a correlated with the severity of diabetic renal damage.11 In addition, patients with DN with C3c deposition in kidney had significantly more severe renal damage than those without C3c deposition.12 These results indicated a pathogenic role of C3a in DN.
Moreover, a broad communication of the C3a with other biological processes including pattern recognition receptors (such as Toll-like receptors and inflammasomes), adaptive immune response and insulin resistance has been reported.13 14 It is interesting that all the aforementioned processes were involved in the pathogenesis of DN,15–17 underscoring a potential role for C3a in regulating these biological processes in DN.
In the current study, the effect of C3a was investigated in a C3aR-deficient (C3aR−/−) diabetic mouse model, and a gene expression microarray was performed to comprehensively explore the underlying mechanism.
Methods
Human renal biopsy samples
Renal tissues were obtained from patients with renal biopsy-proven DN in the Department of Nephrology, Peking University First Hospital. The pathological diagnosis of DN met the criteria proposed by the Renal Pathology Society in 2010.18 Coexistence of other renal diseases such as membranous nephropathy, IgA nephropathy and minimal change disease were excluded. The general data of these patients are shown in online supplementary table S1. Control samples were collected from healthy kidney portions of individuals who underwent tumor nephrectomies without diabetes or renal diseases.
Supplemental material
Animal model
Male C57BL/6J background C3aR knockout and wild-type (WT) mice were generated by Nanjing Biomedical Research Institute of Nanjing University. Mice were housed in the Animal Center of Peking University First Hospital with a 12-hour light–dark cycle allowing free access to food and water. A type 2 diabetic mellitus model was induced according to the previous study with some minor modifications.19 Briefly, 8-week-old mice were fed with high-fat diet (HFD; 60% of total calories from fat, D12492; Keao Xieli, Beijing, China), while the control group was fed with normal chow diet (NCD; 10% of total calories from fat). After 4 weeks, mice in the HFD group were intraperitoneally injected with streptozotocin (STZ, S0130; Sigma-Aldrich, St Louis, USA) dissolved in 50 mM sodium citrate buffer (pH 4.5; Solarbio, Beijing, China) at 60 mg/kg for three consecutive days, while the NCD group were injected with equivalent amount of vehicle. One week after the final STZ injection, fasting blood glucose levels were measured using a glucose analyzer (Accu-check Performa; Roche, Mississauga, ON) on tail-vein blood sample. Only mice with blood glucose levels above 16.7 mmol/L were considered as diabetic. All the mice were sacrificed 20 weeks after STZ induction. All animal experiments were approved by the Laboratory Animal Ethics Committee of Peking University First Hospital (No. 201626).
Urine and blood examination
Mice were placed in a 24-hours metabolic cage for urine collection and urinary albumin levels were measured by a mouse albumin ELISA kit (Bethyl Laboratories, Montgomery, TX, USA). Urinary creatinine levels were assayed by a Creatinine Assay Kit (DICT-500; BioAssay Systems, CA, USA). The ratio of urinary albumin to creatinine was calculated to correct the difference in urine volume and expressed in micrograms per milligram. Before being killed, blood of mice was collected through retro-orbital vein under avertin anesthesia. Plasma cholesterol and triglyceride levels were determined using a UniCel DxC 600 Chemistry Analyzer (Beckman Coulter, CA, USA).
Tissue histology and immunostaining
Four-micrometer-thick, formalin-fixed paraffin-embedded kidney tissue sections were stained with periodic acid–Schiff (PAS, BA-4080B; BASO, Zhuhai, China) solution and glomerular mesangial matrix expansion and tubular injury was investigated as described previously.20 To evaluate the glomerular mesangial matrix expansion, at least 20 glomeruli per renal section were randomly selected and digitized under ×400 magnification and analyzed using Image-Pro Plus software V.6.0 (Media Cybernetics, Bethesda, MD). The mesangial matrix fraction was expressed as the percentage of mesangial matrix expansion area to the glomerular tuft area. To evaluate the tubular injury, all fields were digitized and analyzed under ×200 magnification. Tubular injury was scored from 0 to 5 according to the percentage of damaged tubules (tubular dilation, vacuolar degeneration, interstitial inflammation and cast formation): 0, normal; 1, lesion <10%; 2, 10–20% lesion; 3, 20–30% lesion; 4, 30–40% lesion; 5, >40% lesion.
For immunostaining of C3aR, paraffin-embedded renal sections were subjected to heating in citrated buffer in microwave for antigen retrieval. T lymphocytes and macrophages staining were performed using frozen renal sections. Renal tissues were treated with 3% H2O2, blocked by 3% bovine serum albumin and stained with anti-C3aR antibody (sc-133172; Santa Cruz, CA, USA), anti-CD4 antibody (553043; BD Pharmingen, NJ, USA), anti-CD8 antibody (14-0081-82; eBioscience, MA, USA) and anti-F4/80 antibody (MCA497GA; Bio-Rad, CA, USA) overnight at 4°C. Then anti-mouse and anti-rat secondary antibody (PV-9002, PV-9004; ZSBIO, Beijing, China) were used, followed by color development using 3, 3′-diaminobenzidine (ZLI-9018; ZSBIO). The images were analyzed by Image-Pro Plus under ×200 magnification. Integral optical density was used to represent the expression of C3aR in kidney.
For immunofluorescence staining, anti-human C3aR antibody (sc-133172; Santa Cruz) and anti-CD68 antibody (EPR20545 and ab-125212; Abcam, Cambridge, UK) were used.
Transmission electron microscopy
The renal cortical tissues of mice were cut into three 1 mm3 cubes and immediately immersed into 3% glutaraldehyde after being killed. Further sample handling was performed by the Laboratory of Electron Microscopy, Peking University First Hospital. Two glomeruli were randomly selected from each renal sample and a total of 15–20 representative non-overlapping digital micrographs from each glomerulus were taken on an electron microscope (Hitachi 7700 transmission, Tokyo) under ×10 000 magnification. The number of foot processes per micrometer of glomerular basement membrane (GBM) and GBM thickness were calculated using Photoshop or ImageJ.
Microarray analysis
RNA was extracted by mirVana RNA Isolation Kit (Applied Biosystems p/n AM1556) from three replicate samples per group following the manufacturer’s instructions. The integrity of the RNA was checked on a Bioanalyzer 2100 (Agilent Technologies, USA). The microarray experiments were performed by Shanghai OEbiotech Corporation (Shanghai, China) using Agilent SurePrint G3 Mouse Gene Expression V.2.0 (8×60K) (Agilent Technologies, CA, USA). GeneSpring software (V.13.1; Agilent Technologies) was employed to normalize raw data with the quantile algorithm. Differentially expressed genes (DEGs) were then identified through a fold change ≥2.0 and a p value <0.05. The p value was adjusted by multiple testing using the Benjamin-Hochberg false discovery rate procedure. Afterwards, bioinformatic analysis including Gene Ontology (GO) analysis, Kyoto encyclopedia of genes and genomes (KEGG) analysis and Hierarchical Clustering were applied to display the roles of DEGs. Principal component analysis plots can be found in online supplementary figure S1 and table S2. Microarray data sets have been deposited on Gene Expression Omnibus under accession code GSE133598.
Cell cultures
The immortal mouse macrophage cell line RAW264.7 was obtained from Zhong Qiao Xin Zhou Biotechnology (Shanghai, China). Cells were seeded in 12-well plates and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin–streptomycin (Sigma-Aldrich) in a 5% CO2 humidified incubator at 37°C. Cells were divided into three groups including bovine serum albumin (100 µg/mL, bs-1158PC; Bioss, China), advanced glycation end products (AGEs, 100 µg/mL, bs-1158P; Bioss) and AGEs+C3a (200 ng/mL, 8085-C3-025; R&D, MN, USA). When reaching approximately 70% confluence, the medium was replaced by DMEM containing 5% FBS and cells were treated with different stimuli for 24 hours.
RNA extraction and quantitative real-time PCR (qRT-PCR) analysis
Total RNA of renal cortical tissue or cultured cells was extracted using TRIzol reagent (Invitrogen, MA, USA) and reverse transcribed into cDNA with High Capacity cDNA Reverse Transcription Kits (Applied Biosystems, MA, USA) following the manufacturer’s instruction. Then the qRT-PCR analysis was performed using SYBR green Master Mix (Applied Biosystems) carried out in an ABI Prism 7500 sequence detection system (Applied Biosystems). The primer sequences used for the targeted genes detection are listed in online supplementary table S2. Relative gene expression was normalized to 18s rRNA. Data were presented as relative fold change compared with the control group.
Statistical analysis
Data were presented as the mean±SEM. Differences among multiple groups were determined by one-way analysis of variance followed by Tukey’s multiple comparison test using SPSS software V.19.0. Correlations between parameters were analyzed using Spearman’s correlation test. The p value <0.05 was regarded statistically significant.
Results
C3aR deposition was increased in renal biopsies of patients with DN
To study the role of C3aR in human DN, we compared the renal expression of C3aR between patients with DN and non-diabetic controls by immunohistochemical staining. In non-diabetic controls, hardly any C3aR was detected, while significantly upregulated staining of C3aR was detected in renal biopsies of patients with DN (74.55±49.68 vs 10 636.68±3104.35 pixels, p=0.003). In renal biopsies of patients with DN, C3aR mainly expressed in tubulointerstitial area and to a less extent in glomeruli (mainly in sclerotic glomeruli, figure 1A). We further conducted correlation analysis between the renal expression of C3aR and clinicopathological parameters. The renal expression of C3aR was positively correlated with the percentage of glomerulosclerosis, serum creatinine and interstitial fibrosis and tubular atrophy (IFTA) score (Pearson’s r=0.52, p=0.023; Pearson’s r=0.561, p=0.007; Pearson’s r=0.827, p<0.001, respectively; figure 1B–D).

Increased expression of C3aR in renal biopsies of patients with DN. (A) Representative immunohistochemical staining of C3aR from non-diabetic healthy controls and patients with DN, Bar=100 µm. Correlation analysis between renal C3aR expression and (B) the percentage of glomerulosclerosis, (C) serum creatinine, (D) interstitial fibrosis and tubular atrophy. (E) Co-localization of C3aR and macrophage marker CD68 in kidney of patients with DN, Bar=50 µm. DAPI, 4′,6-diamidino-2-phenylindole; DN, diabetic nephropathy; IOD, integral optical density.
Since macrophages are the predominant immune cells infiltrating in the kidney and are important mediators of inflammation in DN,21 we next explored whether C3aR was expressed on macrophages in patients with DN. CD68 is identified as a classical specific surface marker for macrophages. As expected, C3aR co-localized with CD68 in patients with DN by immunofluorescence co-localization (figure 1E).
C3aR deficiency attenuated renal damage in diabetic mice
After induction of STZ combined with HFD, all the WT mice had developed diabetes, displaying significantly higher levels of blood glucose, serum cholesterol and serum triglyceride, while body weight was decreased compared with non-diabetic WT controls. Compared with WT diabetic mice, C3aR−/− diabetic mice had increased body weight and lower levels of serum triglyceride. Both WT and C3aR−/− diabetic mice developed albuminuria 20 weeks after diabetes induction compared with their non-diabetic controls, whereas C3aR−/− diabetic mice exhibited significantly lower levels of albuminuria compared with WT diabetic mice (160.65±19.72 vs 107.78±10.37 µg/mg, p=0.035; table 1).
Physical and biochemical parameters of diabetic mice
In PAS staining, notably increased mesangial matrix expansion and tubulointerstitial injury were detected in diabetic mice compared with non-diabetic controls. The degree of mesangial matrix expansion was significantly reduced in C3aR−/− diabetic mice compared with WT diabetic mice (figure 2A,E). Due to the relatively low dose of STZ in our diabetic mouse model, the tubulointerstitial lesions were less severe. There were not so many clustered inflammatory cells in the interstitial area in our diabetic mouse model. Compared with WT diabetic mice, the tubulointerstitial injury was significantly improved in C3aR−/− diabetic mice (figure 2B,F).
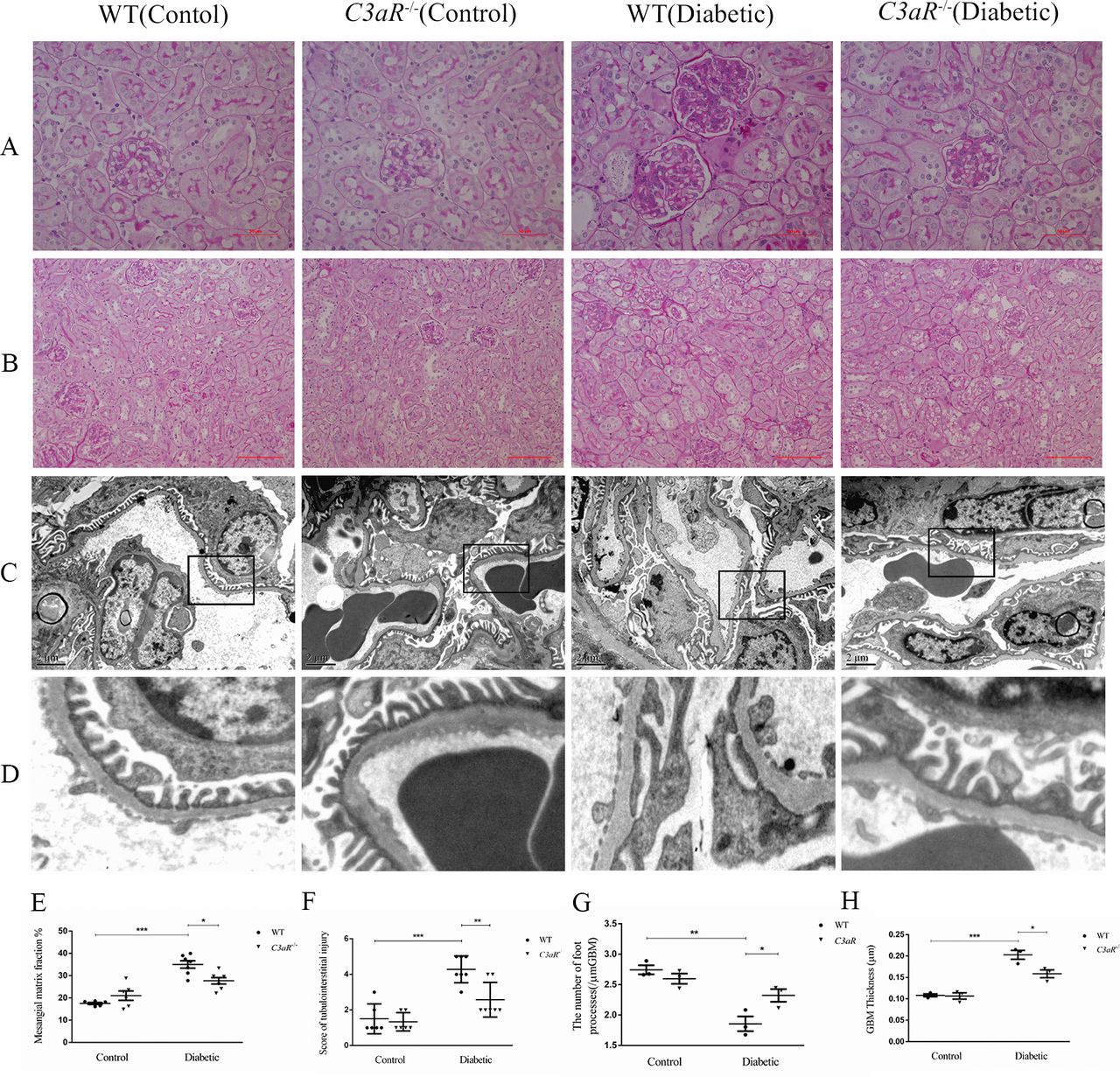
C3aR deficiency attenuated pathological change in diabetic mice. (A) Representative PAS staining of kidney biopsy from non-diabetic controls (n=6) and diabetic mice (n=7) showing mesangial matrix expansion, Bar=50 µm. (B) Representative PAS staining of kidney biopsy from non-diabetic controls and diabetic mice showing tubulointerstitial injury, Bar=100 µm. (C) Representative ultrastructure changes of podocyte and GBM by electron microscopy under ×10 000 magnification, n=3 for each group. (D) Partially enlarged view of electron microscopy. (E) Semiquantitative analysis of mesangial matrix expansion. (F) Semiquantitative analysis of tubulointerstitial injury. Semiquantitative analysis of (G) the number of foot processes and (H) GBM thickness. ***p<0.001; **p<0.01; *p<0.05. GBM, glomerular basement membrane; PAS, periodic acid–Schiff; WT, wild type.
By electron microscopy, we found that diabetic mice exhibited diffused podocyte effacement and thickening of GBM compared with control mice, while C3aR−/− diabetic mice exhibited a significant improvement in the aforementioned ultrastructural abnormalities compared with WT diabetic mice (figure 2C,D,G,H).
C3aR deficiency attenuated diabetic renal damage through suppressing inflammatory responses and T-cell adaptive immune response by microarray
To explore the underlying mechanism of C3aR in renal damage, we performed Agilent Mouse Gene Expression Microarray for global gene expression. By microarray analysis, the C3aR−/− diabetic mice have a total of 104 DEGs changed with a p value <0.05 and fold change ≥2.0 compared with WT diabetic mice. Of these, 96 genes were downregulated and 8 genes were upregulated. KEGG pathway and GO annotation databases were used to provide insight into the potential function and signal pathways of DEGs. The top 10 pathways were cytokine–cytokine receptor interaction, T-cell receptor signal pathway, Th1 and Th2 cell differentiation, hematopoietic cell lineage, Th17 cell differentiation, primary immunodeficiency, chemokine signaling pathway, NF-κB signaling pathway, natural killer cell mediated cytotoxicity and measles (figure 3A). The top 10 biological processes include immune response, immune system process, chemotaxis, adaptive immune response, lymph node development, cell surface receptor signaling pathway, signal transduction, inflammatory response, innate immune response and positive regulation of neutrophil chemotaxis (figure 3B). Both analyses highlighted the regulation of inflammatory responses and T-cell adaptive immunity.
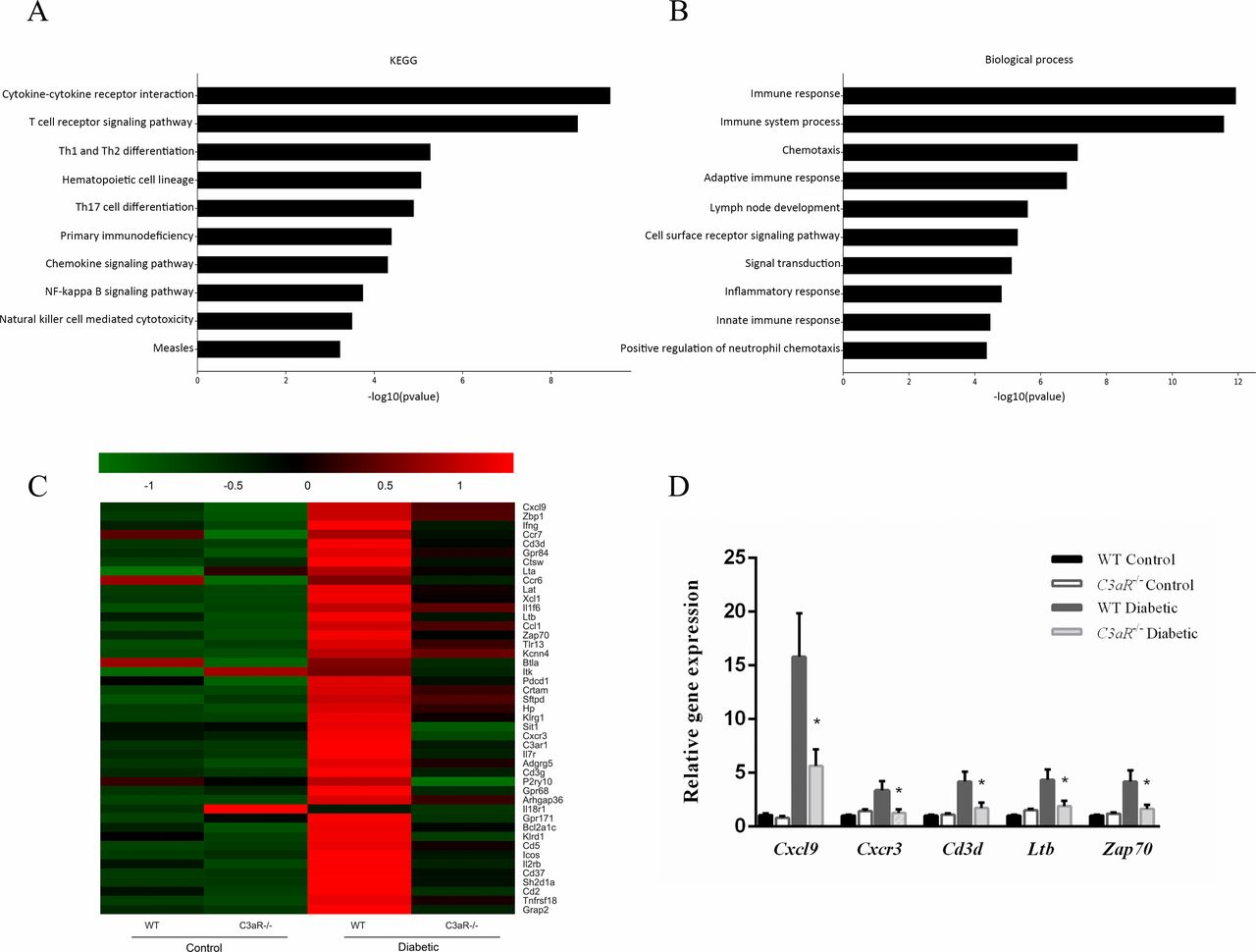
C3aR deficiency attenuated diabetic renal damage through suppressing inflammatory responses and T-cell adaptive immune response by microarray analysis. (A) The top 10 downregulated KEGG pathways of the differentially expressed genes in C3aR−/− diabetic mice compared with WT diabetic mice by p value. (B) The top 10 downregulated biological processes of the differentially expressed genes in C3aR−/− diabetic mice compared with WT diabetic mice by p value. (C) Representative heat map of genes involved in the top 10 KEGG pathways or biological processes. (D) Validation of 5 randomly selected differentially expressed genes by quantitative real-time PCR. *p<0.05 compared with WT diabetic mice. KEGG, Kyoto Encyclopedia of Genes and Genomes; WT, wild type.
A total of 45 genes were involved in the top 10 KEGG pathways or biological processes, which are shown in figure 3C. To validate the results of microarray, five genes including Cxcl9, Cxcr3, Cd3d, Ltb and Zap70 from the 45 genes were randomly selected and qRT-PCR was performed in all mice (figure 3D). The result was consistent with the microarray. These findings indicated that C3aR might exert its effect on DN through influencing inflammation and T-cell adaptive immunity.
C3aR deficiency decreased renal T cells and macrophage infiltration in diabetic mice
Immunostaining identified a twofold to threefold increase of CD4+ and CD8+ T cells in WT diabetic mice compared with non-diabetic controls, while the number of both CD4+ and CD8+ T cells in kidney was significantly reduced in C3aR−/− diabetic mice compared with WT diabetic mice (online supplementary figure S3A–D). By immunostaining of F4/80 which is a specific surface marker of macrophages, we detected macrophage accumulation both in glomeruli and interstitium. Compared with non-diabetic controls, the number of macrophages was remarkably increased in WT diabetic mice and this was diminished in C3aR−/− diabetic kidneys (online supplementary figure S3E, F).
Supplemental material
C3a can enhance the macrophage-secreted cytokines
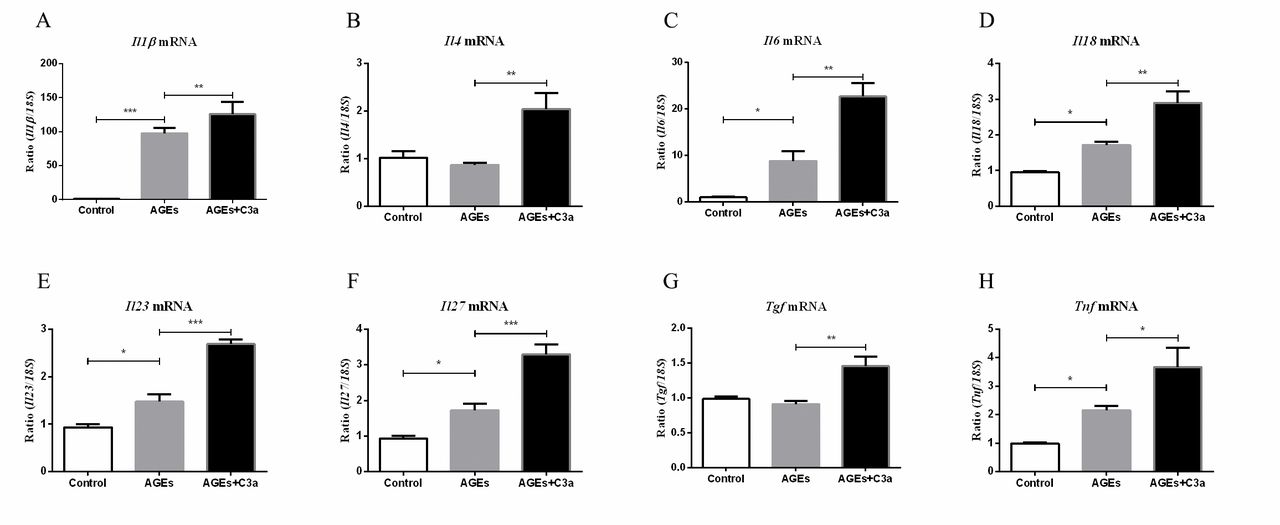
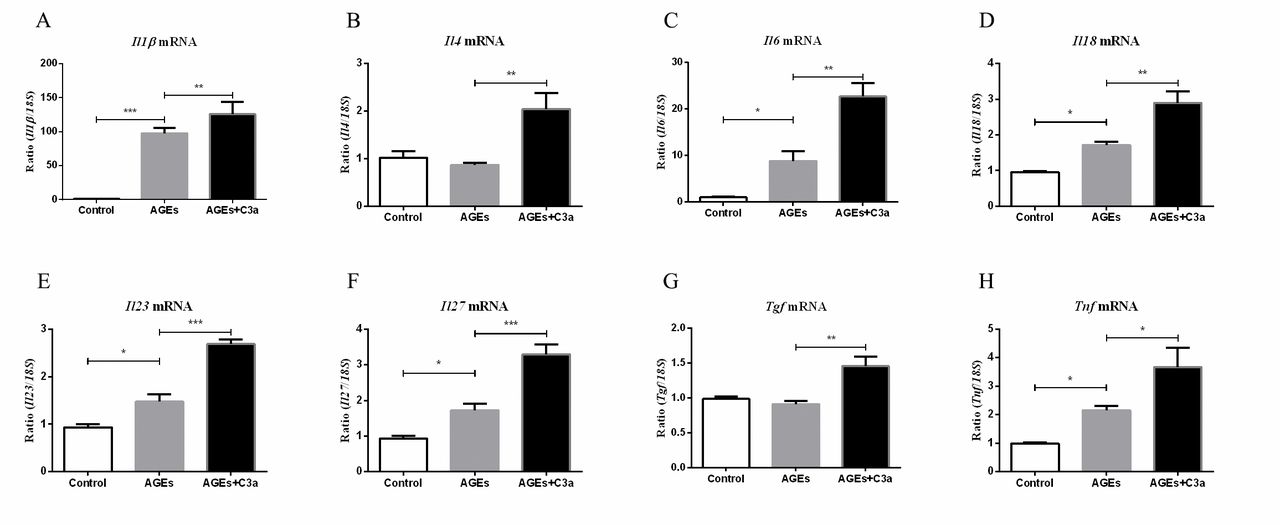
The fate of T cells is shaped by the microenvironment, composed of cytokines, tissue-specific signals and chemokines22; in DN, macrophages are important sources of inflammatory cytokines. Therefore, we investigated the role of C3a on macrophage cell line Raw264.7 of certain cytokines’ expression in vitro. As shown in figure 4, the mRNA expression of Il1β, Il6, Il18, Il23, Il27 and Tnf were significantly upregulated under AGE stimulation compared with the control, and this effect was further enhanced by C3a. Although the levels of Il4 and Tgf did not obviously change under AGE stimulation compared with control, additional of C3a could increase their expression.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
C3a can enhance the macrophage-secreted cytokines. Real-time qPCR analysis of (A) Il1β, (B) Il4, (C) Il6, (D) Il18, (E) Il23, (F) Il27, (G) Tgf and (H) Tnf expression in RAW264.7 (n=5). ***p<0.001; **p<0.01; *p<0.05. AGEs, advanced glycation end products.
Discussion
In the past decades, a number of evidence revealed a role of the complement system in DN. To be more specific, two main mechanisms are thought to be involved in the pathogenesis of DN. First, under hyperglycemia, enhanced glycated proteins such as fructosamines would be recognized by elevated level of mannose-binding lectin, resulting in the activation of lectin pathway.23 Second, hyperglycemia is also thought to induce glycation of complement regulatory proteins,24 25 breaking the subtle balance between complement activation and restriction, leading to overactivation of the complement system. Li et al have previously reported that C3a could aggravate diabetic kidney injury through acting on renal glomerular endothelial cells by using a C3aR inhibitor26 27; however, the underlying mechanism still needs further investigation.
In our previous study, it was found that both plasma and urinary C3a levels were significantly increased in patients with DN, and the urinary levels of C3a correlated with the severity of diabetic renal damage.11 In the current study, we found that in renal biopsy of patients with DN, the expression of the C3aR was significantly higher compared with non-diabetic controls, and the renal expression of C3aR was positively correlated with the severity of diabetic renal lesions, including percentage of glomerulosclerosis, serum creatinine and IFTA score. By gene knockout of C3aR in diabetic mouse model, we showed that C3aR−/− diabetic mice exhibited significantly lower levels of albuminuria and milder pathological changes compared with WT diabetic mice. Microarray was performed to further explore the underlying molecular mechanism. It turned out that, by bioinformatics analysis, the inflammatory response and T-cell adaptive immunity were restrained in C3aR−/− diabetic mice, as compared with WT diabetic mice.
Cumulating evidence highlights the critical role of inflammation in the development of DN,28 in which macrophages are the main mediator.29 Macrophages are the most prevalent infiltrating leucocytes and are important sources of proinflammatory cytokines in DN. The accumulation of macrophages in kidneys of patients with DN correlated with declining renal function.21 Our current study found that C3aR co-localized with renal macrophages; under C3a stimulation, the mRNA expression of inflammatory cytokine such as Il1β, Il6, Il18 and Tnf were significantly increased in macrophages. It is therefore possible that C3aR deficiency attenuated diabetic renal damage through alleviating local inflammation by reducing the cytokine production by macrophages.
Although DN was originally regarded as an innate immunity–mediated rather than adaptive immunity–mediated disease, recently growing evidence indicated adaptive immunity, mainly T-cell immunity, was also involved in pathogenesis of DN.30 Increased renal T-cell recruitment were detected both in patients with DN and diabetic mice.31 A series of investigations inhibiting T-cell activation or targeting Th17 ameliorated diabetic renal damage in animal models.32–35 In our study, we found the T-cell immune response was suppressed in C3aR−/− diabetic mice compared with WT diabetic mice by microarray, indicating a link between C3a and T-cell adaptive immunity in DN. It is well known that the cytokine microenvironment could influence T-cell response.36 Our study also found that under C3a stimulation, the mRNA expression of Il4, Il23, Il27 and Tgf of macrophages was upregulated. This might result in the differentially modulated T-cell response in diabetic mice. Except for the indirect effect through macrophages, C3a also has a direct effect on T cell since intracellular complement activation sustains T-cell homeostasis and mediates effector differentiation.37 We speculated that this effect also plays a role in our C3aR−/− diabetic mice.
Compared with the studies by Li et al,26 27 the current study further extended the role of C3a in DN by gene knockout of C3aR in diabetic mice model, and microarray was performed to further investigate the function of C3a in DN. Besides, we revealed the important effect of C3a on macrophage in DN.
There were several limitations of the study. First, although the current study mainly investigated the role of C3a on macrophage, we could not exclude the effect of C3a on tubular cells and glomerular cells. Future study of macrophage-specific C3aR knockout rather than the global C3aR knockout mice is needed to further elucidate this question. Second, since the microarray analysis was performed in three animals per group, this may limit the interpretation of the results due to the variation of the samples.
In conclusion, C3aR deficiency could attenuate diabetic renal damage through suppressing inflammatory responses and T-cell adaptive immunity, and these effects were possibly mediated by influencing macrophage-secreted cytokines. Thus, C3a might be a bridge linking innate immunity and adaptive immunity in DN, and it might be a promising therapeutic target for DN.
Supplemental material
Supplemental material
References
Footnotes
Contributors MC designed the study. D-YC collected the renal biopsies. X-QL carried out the experiments, analyzed the data, made the figures and drafted the paper. MC and M-HZ revised the paper. All authors approved the final version of the manuscript.
Funding This study was supported by the grants from the National Key Research and Development Program (No. 2016YFC1305405), two grants from the National Natural Science Fund (Nos. 81425008 and 81621092), the grant by Peking University Health Science Center (No. BMU2017CJ002), and a grant from the University of Michigan Health System and Peking University Health Sciences Center Joint Institute for Translational and Clinical Research.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The study was approved by the Research Ethics Committee of Peking University First Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as online supplementary information.