Article Text

Abstract
Introduction Diabetes is a worldwide health problem that is associated with severe complications. Advanced Glycation End products (AGEs) such as Nε-(carboxymethyl)lysine, which result from chronic hyperglycemia, accumulate in the skin of patients with diabetes. The effect of AGEs on fibroblast functionality and their impact on wound healing are still poorly understood.
Research design and methods To investigate this, we treated cultured human fibroblasts with 0.6 mM glyoxal to induce acute glycation. The behavior of fibroblasts was analyzed by time-lapse monolayer wounding healing assay, seahorse technology and atomic force microscopy. Production of extracellular matrix was studied by transmission electronic microscopy and western blot. Lipid metabolism was investigated by staining of lipid droplets (LDs) with BODIPY 493/503.
Results We found that the proliferative and migratory capacities of the cells were greatly reduced by glycation, which could be explained by an increase in fibroblast tensile strength. Measurement of the cellular energy balance did not indicate that there was a change in the rate of oxygen consumption of the fibroblasts. Assessment of collagen I revealed that glyoxal did not influence type I collagen secretion although it did disrupt collagen I maturation and it prevented its deposition in the extracellular matrix. We noted a pronounced increase in the number of LDs after glyoxal treatment. AMPK phosphorylation was reduced by glyoxal treatment but it was not responsible for the accumulation of LDs.
Conclusion Glyoxal promotes a change in fibroblast behavior in favor of lipogenic activity that could be involved in delaying wound healing.
- fibroblasts
- collagen
- diabetic foot
- skin
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Advanced Glycation End products (AGEs) accumulate over time in the skin of patients with diabetes.
Collagen is one of the first targets of glycation, and this leads to the disruption of the dermis, thereby potentially contributing to the skin complications in diabetes.
The accumulation of AGEs leads to impaired wound healing and the formation of chronic ulcers.
What are the new findings?
There have been few studies to date of the phenotype of dermal fibroblasts in the presence of glyoxal, and our research shows that AGEs alter key properties of fibroblasts.
Exposure to glyoxal reduced fibroblast proliferation and migration.
We demonstrate that although collagen was being synthesized, it was not being deposited in the extracellular matrix.
For the first time, we show that glycation can induce ectopic lipid accumulation through increased expression of PLIN2 in dermal fibroblasts, independently of the AMPK pathway.
How might these results change the focus of research or clinical practice?
Treatment of fibroblasts with glyoxal reproduces some of the modifications found in the skin of diabetics.
This model could be used to study the impaired wound healing in patients with diabetes and to test new therapeutic molecules and approaches.
Introduction
Diabetes is a metabolic disease and a major public health problem: it affected more than 460 million people in the world in 2019 (International Diabetes Federation). Chronic hyperglycemia is the main feature of type I and type II diabetes, which leads to microvascular, neuropathic, and skin complications. Indeed, 6.3% of diabetics suffer from a substantial delay in wound healing, which results in chronic ulcers that can lead to foot amputation. Approximately one million patients with diabetes undergo toe or foot amputations every year.1
Advanced glycation end products (AGEs) are one of the key factors involved in hyperglycemia-related complications. AGEs are the result of the chemical reaction of glucose with protein amino acid residues, which then initiates a complex process through a non-enzymatic reaction called the Maillard reaction.2 3 Common reaction products are members of the α-oxoaldehyde family, which comprise glyoxal, methylglyoxal, and 3-deoxyglucosone (3-DG).2 Mera et al reported that the formation of AGEs in vivo is highly dependent on α-oxoaldehyde and may contribute to diabetic complications.4 Glyoxal is found in large quantities in the plasma of patients with diabetes and it is responsible for the formation of Nε-(carboxymethyl)lysine (CML).3 5–7 Proteins with a slow turnover rate such as collagen are particularly subject to glycation and can be cross-linked by glyoxal.5 Thus, diabetes results in the accumulation of AGEs in skin as collagen-CML which may contribute to the disruption of skin integrity, as indicated by the high rate of impaired wound healing in patients with diabetes.8–10 A wound that do not heal after 3 months is called a chronic wound. This generally occurs when the inflammatory, the proliferative, and the remodeling phases of the wound healing process are prolonged and/or altered.11
The proliferation phase involves fibroblasts from the non-injured areas that start to proliferate and migrate towards the center of the wound.12 13 At this time, the fibroblasts secrete mainly collagen III and they can differentiate into myofibroblasts that have contractile capacities to reach the wound edges. At the same time, neoangiogenesis results in vascularization of the dermis, and re-epithelialization closes the epidermis. Then, the purpose of the remodeling phase of the extracellular matrix (ECM) is to replace the scar tissue with a physiological matrix. During this phase, the fibroblasts produce mainly collagen I in order to restore a matrix that resembles uninjured tissue.14 Here, we wanted to study the behavior of fibroblasts in a context of increased glycation, as is the case in patients with diabetes. Many studies have shown that α-oxoaldehyde-modified collagen affects fibroblast viability.15 16 However, the neosynthesis of collagen by fibroblasts and their capacities to migrate and proliferate in the presence of AGEs remain unclear.
In addition, it is known that glyoxal can mimic the cellular metabolic stresses in diabetes.17 Indeed, diabetes is linked with ectopic fat accumulation in non-adipose tissue, including skeletal muscle and liver, associated with insulin resistance and cell dysfunction.18 19 An increase in lipid droplets (LDs) and their associated protein perilipin-2 (PLIN2) are associated with metabolic diseases such as diabetes or obesity.20 21 Here, we investigated whether glycation by glyoxal could result in impaired lipid metabolism, with an accumulation of LDs, as observed in patients with diabetes.
The aim of this study was to investigate how increased glycation can affect the dermis during diabetes in order to obtain a better understanding of the impaired wound healing in patients with diabetes. We focused on the behavior of fibroblasts treated with an acute dose of glyoxal to mimic the accumulation of AGEs that occurs with diabetes. Our results could be used to study related skin diabetic complications and test new therapeutic molecules for their ability to improve skin healing.
Research design and methods
Cell culture
Normal human dermal fibroblasts were obtained from breast biopsies of women who were between 18 and 23 years of age and who had signed an informed consent form. One biopsy was provided by the ‘Hospice civils de Lyon’ (DC-2008–162 delivered to the Cell and Tissue Bank of Hospices Civils de Lyon), while the other two cell samples were generated by PromoCell (Lot numbers #401Z028.2 and  #428Z014.2). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) low glucose (1 g/L) (Thermo Fisher Scientific, #31885049) supplemented with 1% ZellShield (Minerva Biolabs, #130050) and 10% fetal calf serum (FCS) at 37°C in a 5% CO2 atmosphere, and the cells were used between the fourth and the eighth passage. To study the ECM, 50 µg/mL of ascorbic acid was added once the cells had reached confluency.
#428Z014.2). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) low glucose (1 g/L) (Thermo Fisher Scientific, #31885049) supplemented with 1% ZellShield (Minerva Biolabs, #130050) and 10% fetal calf serum (FCS) at 37°C in a 5% CO2 atmosphere, and the cells were used between the fourth and the eighth passage. To study the ECM, 50 µg/mL of ascorbic acid was added once the cells had reached confluency.
Flow cytometry
Flasks (75 cm2) were seeded with 3×105 cells, and after 3 days, 0.6 mM glyoxal (Sigma-Aldrich, #50649) was added to the proliferating fibroblasts for 24 hours. After trypsinization and centrifugation, the cells were resuspended in 500 µL (2×106 cells per mL) of binding buffer (10 mM HEPES, 140 mM NaCl, 4 mM KCl, and 0.75 mM MgCl2). A working solution of 2.5 mM CaCl2, 5 µL Annexin V-APC (BD Biosciences #550474), and 2.5 µg/mL propidium iodide (Roche # 11348639001) was added to 100 µL of cell suspension and incubated for 10 min on ice in the dark. The stained cells were subjected to flow cytometry using a BD Biosciences FACS Melody, using a minimum of 30 000 cells for each sample, and the flow cytometry signal was analyzed with Flow Jo software. Annexin V-/ PI- cells were considered to be non-apoptotic and viable, Annexin V+/PI+ cells were considered to be in late apoptosis, and Annexin V+/PI- cells were considered to be in early apoptosis.
Proliferation test
Fibroblasts (9.5×103) were seeded into 6-well sterile plates. After 24 hours, 0.6 mM glyoxal was added for a further 24 hours and the cells were counted using a Malassez counting chamber over the course of 5 days in the presence of trypan blue.
Time-lapse monolayer wounding healing assay
Fibroblasts (5.7×104) were seeded into 24-well sterile plates and cultured until confluent. Monolayer wound healing assays were performed by scraping the confluent cells, as previously described by Viglio et al, with a 20 µL pipette tip.22 DMEM low glucose with 1% FCS was added to reduce cell proliferation, with or without 0.6 mM glyoxal. Pictures of the monolayer wounds were taken every 10 min for 48 hours using an inverted microscope at 37°C and 5% CO2. Image J software was used to measure the area not covered by cells (the non-healed area) every 6 hours from 0 hours to 48 hours. The percentage monolayer wound healing was quantified by calculating:  .
.
Atomic force microscopy
Fibroblasts (16×104) were seeded into 35 mm Petri dishes. After 1 day, ascorbic acid (50 µg/mL) was added for another 4 days in order to produce collagen fibers, and 0.6 mM glyoxal was then added for a further 24 hours. The cells were then fixed with 4% paraformaldehyde for 15 min. The subsequent measurements and analyses were carried out by BioMeca SAS (Lyon, France). A Bioscope Resolve device (Bruker) in PeakForce QNM (Quantitative Nanomechanical Mapping) mode was used for the atomic force microscopy (AFM), with a probe that had a stiffness constant of 0.35 N/m and a radius of curvature <10 nm. The force measurements were performed in 1x Phosphate-Buffered Saline (PBS). Acquisition of the force volume of 10 μm × 10 μm was performed on the collagen at the edge of the fibroblasts. Each force volume included 4096 measurement points. Each measurement point corresponded to an indentation force curve, from which the elastic modulus (Ea) was then extracted. Quantification of the elastic modulus from the raw force curves was performed using BioMeca Analysis processing software.
Extracellular matrix and supernatant protein extraction for determination of collagen I expression
Fibroblasts (1.68×106) were seeded and cultured in 100 mm Petri dishes until confluent and then treated with 50 µg/mL ascorbic acid, with or without 0.6 mM glyoxal. Six days later, the cells were washed with PBS and FCS-free medium was added. The medium was collected 2 days later.
The proteins in the medium were precipitated with 0.02% Triton X-100% and 35% trichloroacetic acid and incubated for 20 min on ice. The samples were centrifuged at 19 000× g for 20 min at 4°C, and the supernatants were removed. The pellets were washed with cold absolute ethanol. After centrifugation at 19 000× g for 10 min at 4°C, the pellets were washed with cold acetone. Finally, after another centrifugation at 19 000× g for 10 min at 4°C, the pellets were resuspended in Radioimmunoprecipitation Assay (RIPA) buffer containing a protease inhibitor cocktail.
In order to extract proteins from the cells and the ECM, extraction buffer (0.5 M Na2HPO4 pH 7.7, 8 M Urea, 10% NP-40, protease and phosphatase inhibitors, and 0.2 M EDTA) was then added to the Petri dishes for 2 hours at 4°C. The proteins were diluted fourfold in 0.5 M Na2HPO4, 0.2 M EDTA containing protease and phosphatase inhibitors. After centrifugation at 20 000× g for 5 min at 4°C, the supernatants were removed and the proteins precipitated with 10% trichloroacetic acid. The pellets were washed with cold acetone after centrifugation at 10 000× g for 10 min at 4°C and then resuspended in RIPA buffer (Sigma #R0278) containing a protease inhibitor cocktail (Merck #5056489001).
Intracellular protein extraction for determination of AMPK expression
Fibroblasts (5.6×105) were seeded into 100 mm Petri dishes and after 24 hours, 0.6 mM glyoxal was added for a further 24 hours. The cells were lysed with Tissue Protein Extraction Reagent (T-PER) buffer (Thermo Fisher Scientific, #78510) containing a proteases inhibitor cocktail (Merck, #5056489001) and phosphatase inhibitors (5 mM NaF, 10 mM β-glycerol phosphate, and 1 mM Na3VO4) for 30 min at 4°C. After centrifugation at 14 000× g, the supernatants were collected.
Western blotting
The total protein concentration was determined using a Pierce BCA Assay Kit (Thermo Fisher Scientific, #23225) according to the manufacturer’s recommendations. The absorbance at 560 nm was measured. The proteins were denatured for 5 min at 95°C in Laemmli buffer (62.5 mM Tris-HCl pH 6.8, 0.01% bromophenol blue, 10% glycerol, 2% sodium dodecyl sulfate (SDS), and 50 mM 1,4-dithiothreitol (DTT), separated by polyacrylamide gel electrophoresis according to the protein size in 1× tris-glycine SDS buffer. The proteins were then electroblotted onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore, #IPVH00010) in TG-20% ethanol for between 1h30 and 3h00 at 220 mA at 4°C. The PVDF membranes were then blocked for 1 hour with 1× tris-buffered saline (TBS) containing 0.1% Tween 20% and 5% nonfat milk or bovine serum albumin (BSA) depending on the antibodies to be used. The primary antibodies were diluted in 1× TBS containing 0.1% Tween and nonfat milk (2%) or BSA (5%) and incubated overnight at 4°C: rabbit polyclonal anti-type I collagen (Novotec, #20111, 1:1000), rabbit polyclonal anti-carboxymethyl-lysine (Abcam, #ab27684, 1:500), rabbit polyclonal anti-AMPK (cell signaling #2532, 1:1000), rabbit monoclonal anti-P-AMPK (Thr172) (cell signaling #2535, 1:1000), mouse monoclonal anti-actin (Merck, #6A2910, 1:5000). The membranes were incubated for 1 hour at room temperature with goat anti-rabbit HRP conjugate (BioRad #1706515) or goat anti-mouse HRP conjugate (BioRad #1706516) diluted 1:5000 in 1× TBS, 0.1% Tween 20. The membranes were then incubated for 5 min with ECL reagent (Thermo Fisher Scientific, #34580), and the antigens detected by chemiluminescence. The signal was recorded using a Fusion Fx system. Protein signals were evaluated using GelAnalyzer software.
Indirect immunofluorescence staining
Fibroblasts (3.8×104) were seeded onto 12 mm coverslips. After 24 hours, 0.6 mM glyoxal, 1 mM AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) (Sigma #A9978), or both, were added for a further 24 hours and the cells were then fixed with 2% formaldehyde for 20 min. BODIPY 493/503 (Thermo Fisher Scientific, #D3922) and anti-perilipin-2 staining (Santa Cruz Biotechnology, #sc-377429) were performed according to the protocol of Listenberger et al.23 The coverslips were then incubated for 1 hour at room temperature with Alexa Fluor 488-conjugated anti-rabbit IgG secondary antibodies (1:1000) (Thermo Fisher Scientific, #A11034). The nuclei were labeled with 2 µg/mL 4',6-diaminido-2-phenylindole (DAPI) and the coverslips were then mounted with PermaFluor (Thermo Fisher Scientific, #TA-030-FM). Images were acquired with a Nikon ECLIPSE Ti-E microscope and NIS-Element software.
Transmission electron microscopy
Fibroblasts (2.7×106) were seeded into 35 mm Petri dishes and cultured until confluent, followed by treatment with 50 µg/mL ascorbic acid, with or without 0.6 mM glyoxal, for a further 8 days. The cells were fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.3 for 1 hour at 37°C. The cells were then rinsed three times with 0.2 M sodium cacodylate buffer pH 7.3 and stored at 4°C. All of the samples were postfixed in 1% osmium tetroxide for 1 hour at room temperature and embedded in epoxy resin (50% epon A/50% epon B). Ultrathin sections (80 nm) were contrasted with 7% uranyl acetate and lead citrate and examined using a Philips CM 120 electron microscope (Centre Technologique des Microstructures, Université Claude Bernard Lyon 1, Villeurbanne, France).
Seahorse extracellular flux analysis of mitochondrial respiration
Cells (4×104) were seeded on a Seahorse XFe24 microplate (Agilent, #1 00 777–004) and after 24 hours 0.6 mM glyoxal was added for a further 24 hours. A Seahorse XFe24 Sensor Cartridge (Agilent, #1 02 340–100) was incubated with XF calibrant (Agilent, #1 03 059–000) solution overnight at 37°C in a non-CO2 humidified incubator. Seahorse XF DMEM assay medium pH 7.4 (Agilent, #1 03 575–100) was complemented the day of the experiment with 10 mM glucose (Agilent, #1 03 577–100), 1 mM pyruvate (Agilent, #1 03 578–100), and 2 mM L-glutamine (Agilent, #1 03 579–100). The cells were then washed with assay medium and placed for 1 hour in a non-CO2 humidified incubator. Just prior to the assays, the cells were washed again. The assays were performed with an Agilent Seahorse XFe24 analyzer with controller after calibration of the cartridge. Oligomycin (1.5 µM), Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) (3 µM), and rotenone/antimycin A (1 µM) were then added sequentially with three cycles of measurement (mix 3 min, wait 2 min, and measure 3 min). Seahorse Wave software was used to analyze the Seahorse measurements.
Statistical analysis
The results are expressed as means±SD. The statistical tests were performed using GraphPad Prism software. Mann-Whitney or t-tests were used to compare the means of two groups. Multiple t-tests were used to compare two groups independently of other conditions. One-way analysis of variance (ANOVA) was used to compare different groups between each other. Two-way ANOVA with multiple comparisons was used for the intergroup and intragroup comparisons at different time points. Significance was defined as p<0.05.
Results
Glyoxal treatment resulted in CML production but no changes in metabolism or viability
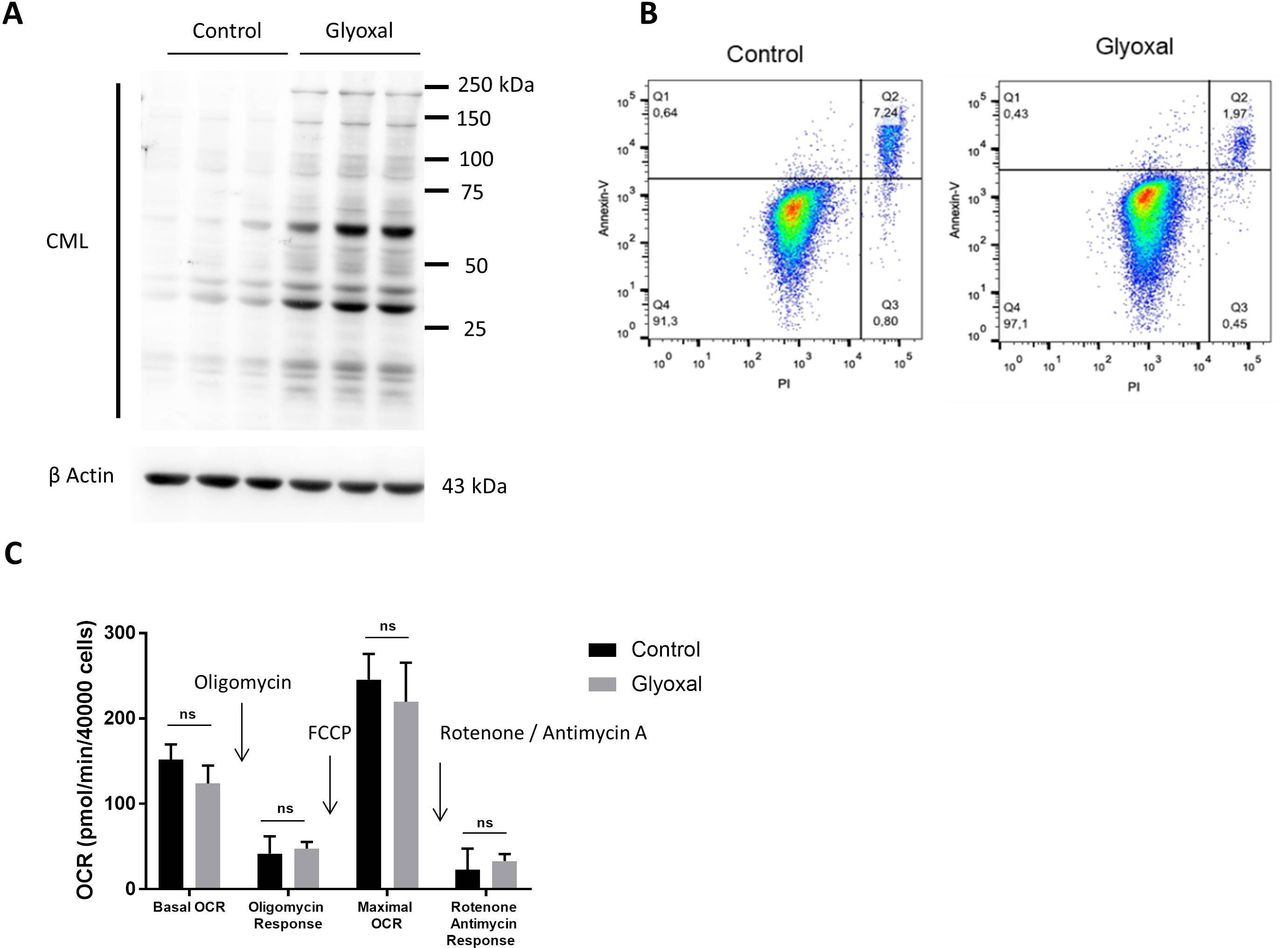
We first of all used Western blotting to confirm the production of CML by fibroblasts after 24 hours of treatment with glyoxal (figure 1A). Flow cytometry assessment of apoptosis was used to verify whether glyoxal exposure induced cell death after a 24 hours of treatment. More than 90% of the cells were negative for annexin V and propidium iodide staining (figure 1B), indicating that glyoxal did not induce a preapoptotic phenotype. The innovative Seahorse technology allowed real-time analysis of cell function by quantification of the oxygen consumption rate. We found that glyoxal exposure did not alter the mitochondrial respiration rate, as the cells were metabolically active after 24 hours of glyoxal treatment (figure 1C). These results showed that 0.6 mM glyoxal induced CML production without affecting the viability or the mitochondrial respiration of the fibroblasts.
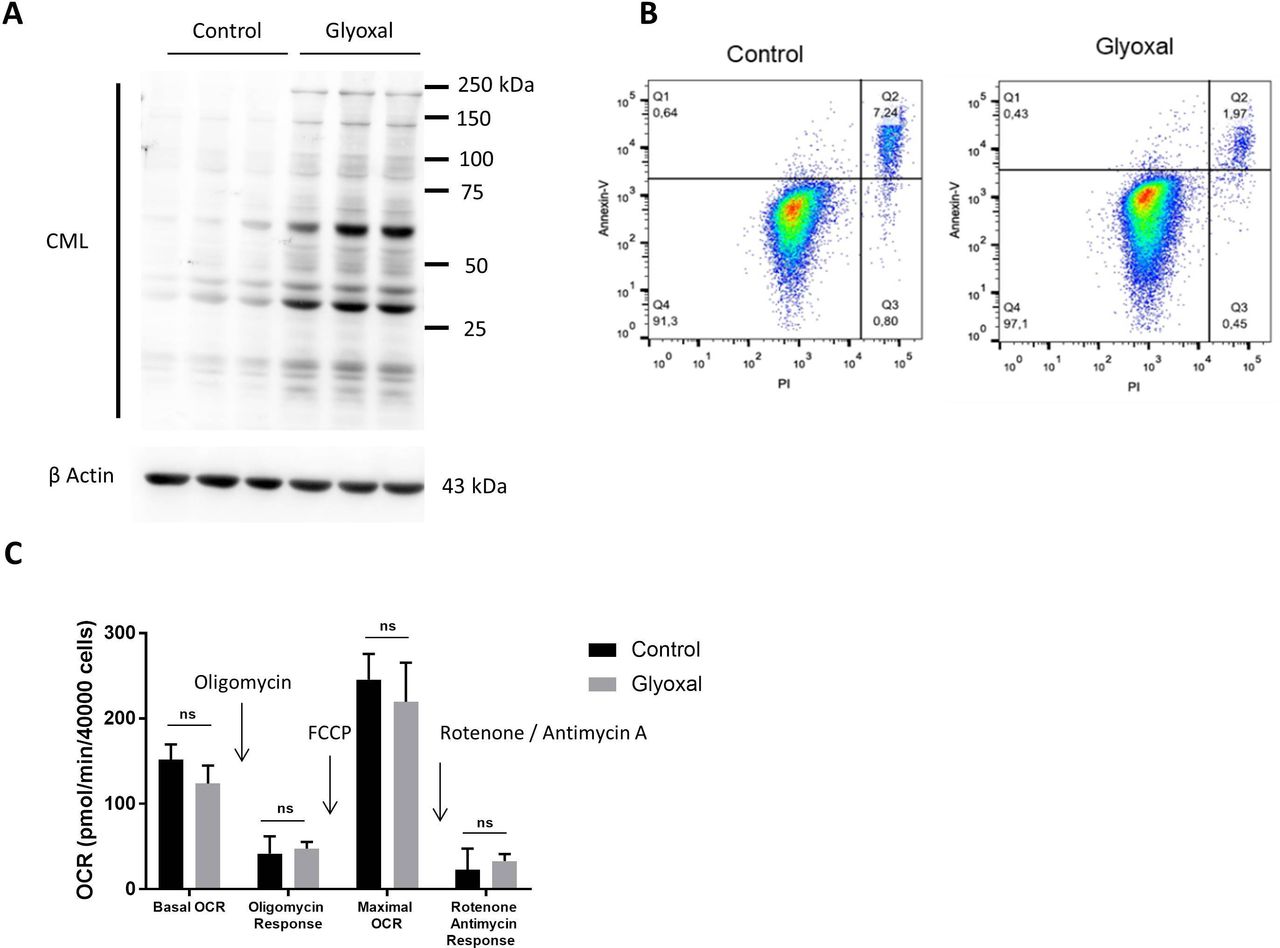
Glyoxal-induced CML production without induction of apoptosis or an effect on mitochondrial respiration. Fibroblasts were treated with or without 0.6 mM glyoxal for 24 hours. Control: untreated cells; glyoxal: 0.6 mM. (A) Western blot analysis of CML and β actin. The assays were performed on fibroblasts from three different donors. (B) Annexin V-APC and PI flow cytometry analysis of the fibroblasts. (C) Oxygen consumption rate measurement using Seahorse technology. Quantification of cell reactivity to drugs. Oligomycin (1.5 µM), FCCP (3 µM), and rotenone/antimycin A (1 µM) were added sequentially. The average of three measurements after drug injection is represented for each treatment. Two-way ANOVA of the control vs glyoxal treatment is indicated, ns=non-significant. The data represent the means±SD. ANOVA, analysis of variance; CML, Nε-(carboxymethyl)lysine; PI, propidium iodide.
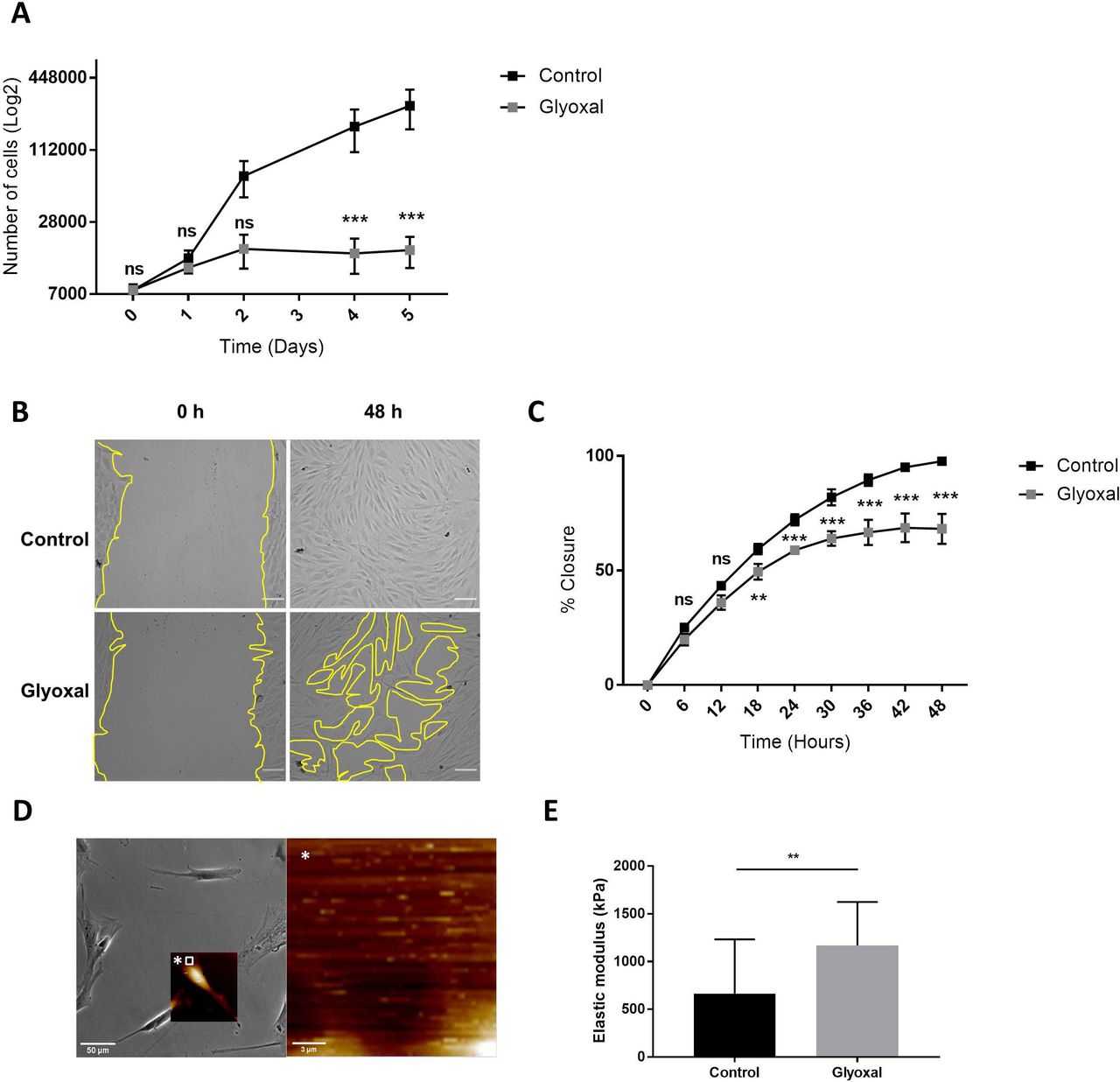
Glyoxal treatment impaired fibroblast proliferation and migration
Next, we examined the impact of glyoxal exposure on cell behavior and particularly on cell proliferation and migration. The proliferation rate of the fibroblasts treated with glyoxal started to decrease at day 2, and it was significantly different from the fourth day until day 5 (figure 2A). We also investigated the migration of the fibroblasts in the presence and absence of glyoxal using an in vitro monolayer wounding healing assay (figure 2B). Compared with the rapid wound healing in the controls, the treated cells required more than 2 days to close the wound (figure 2B,C). The percentage wound closure was significantly reduced from 18 hours up to 48 hours for the cells treated with glyoxal compared with the control cells, and it reached a plateau at 30 hours (figure 2C). In an effort to shed further light on the impaired fibroblast migration in patients with diabetes, we used AFM to analyze the mechanical forces exerted on neo-collagen by cells exposed to glyoxal for 24 hours. To estimate the tensile strength of the fibroblasts, we measured the elastic modulus of the collagen at the edge of cells (figure 2D). This elastic modulus is proportional to the deformation of the collagen network, which stretches in response to the traction exerted by the fibroblasts. We demonstrated that the tensile strength was significantly increased in the presence of glyoxal, going from 500 kPa for the control to approximately 1200 kPa for the glycated cells (figure 2E). An increase in cell contraction was also noted by measurement of the cell area in the presence of glyoxal (online supplemental figure S1). Taken together, these mechanical parameters demonstrate that the glyoxal-treated fibroblasts adhered more strongly to the ECM than the untreated cells.
Supplemental material
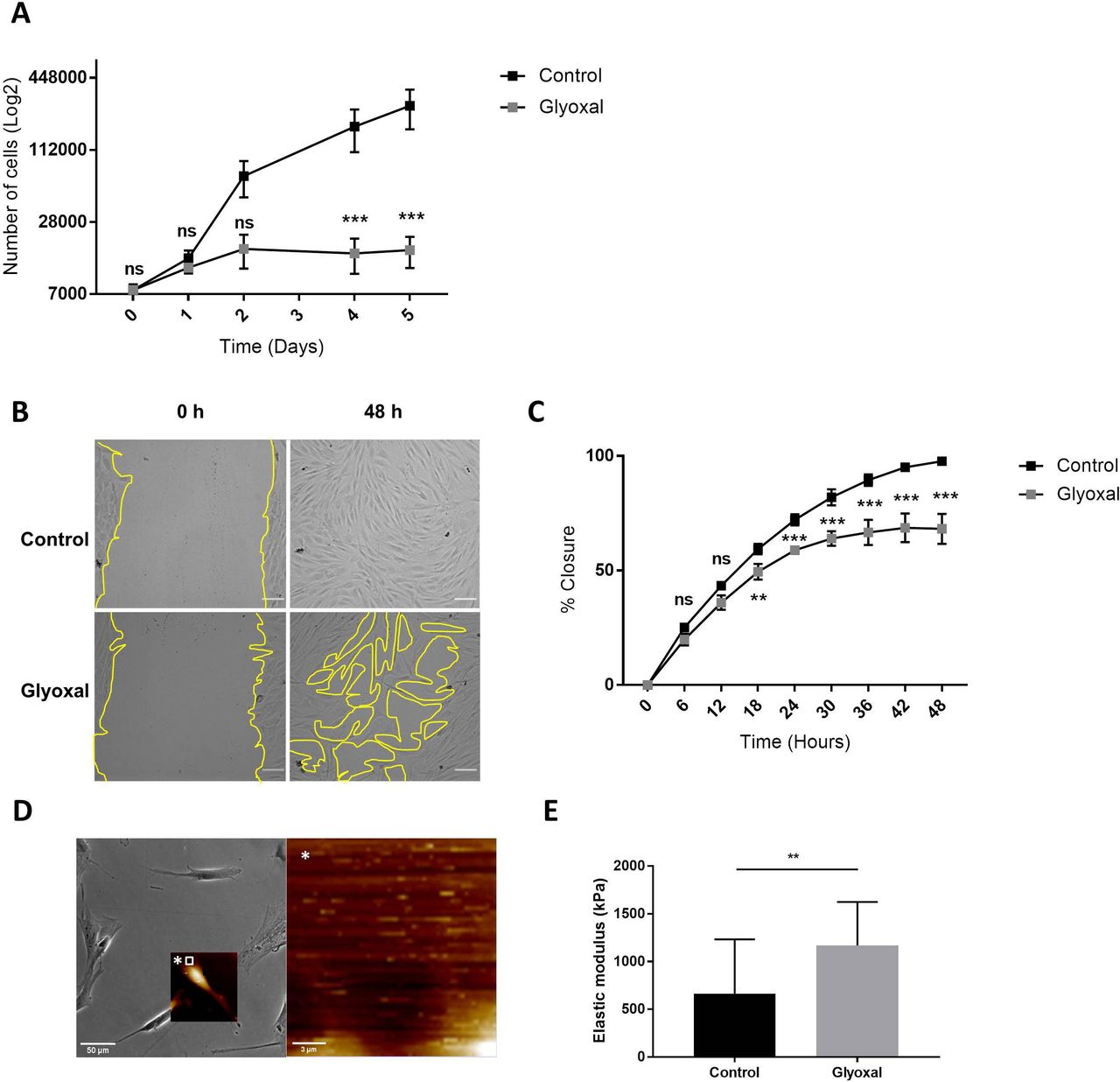
Glyoxal reduced proliferation and migration but enhanced cell contraction. The assays were carried out on fibroblasts from three different donors. Control: untreated cells; glyoxal: 0.6 mM. (A) Proliferation of the fibroblasts was estimated by daily counting of the cells for 5 days. (B) Images of the monolayer wounding at 0 hour (immediately after the monolayer wound was made) and healing 48 hours later in the presence or absence of glyoxal. The yellow lines represent an example of the measured area (scale bare=100 µm). (C) Migration of the fibroblasts, quantified as the percentage wound closure. The unclosed areas were quantified every 6 hours for 48 hours in six different wells for each donor. (D) Example of the location of the elastic modulus measured by AFM (white square) The white star represents a magnification of the white square. (E) The elastic modulus (kPa) of the collagen on the edge of the fibroblasts represents the tensile strength exerted by the cells as measured by AFM in the absence and presence of glyoxal (five fibroblasts in three different wells from one donor). Two-way ANOVA of the control vs glyoxal treatment is indicated in (A,B). A Mann-Whitney test of the control vs glyoxal treatment is indicated in (C). **p<0.01; ***p<0.001; ns=non-significant. The data represent means±SD. AFM, atomic force microscopy; ANOVA, analysis of variance.
Glyoxal treatment did not influence type I collagen secretion, but it did impair collagen I cleavage and maturation and it prevented deposition of extracellular matrix components
To verify whether the dermal fibroblasts were able to produce and secrete ECM components, we used Western blotting to evaluate the amount of type I collagen in the cell supernatants in the presence and absence of glyoxal (figure 3A). There appeared to be no difference between the treated and the untreated cells in terms of the amount of pro-collagen I, thus indicating that the glyoxal treatment did not affect the secretion of type I collagen (figure 3B). Nonetheless, the level of cleaved and matured type I collagen was reduced by the glyoxal treatment (figure 3B). Extraction of the ECM proteins, which yields the deposited forms, revealed the presence of pro-collagen I, mature collagen I, intermediate forms, and cross-linked forms in the controls (figure 3C). Following glyoxal treatment, we detected only a small amount of mature collagen I, but no cross-linked forms (figure 3D), thus indicating that the cells were unable to produce normal collagen under the glycating conditions.
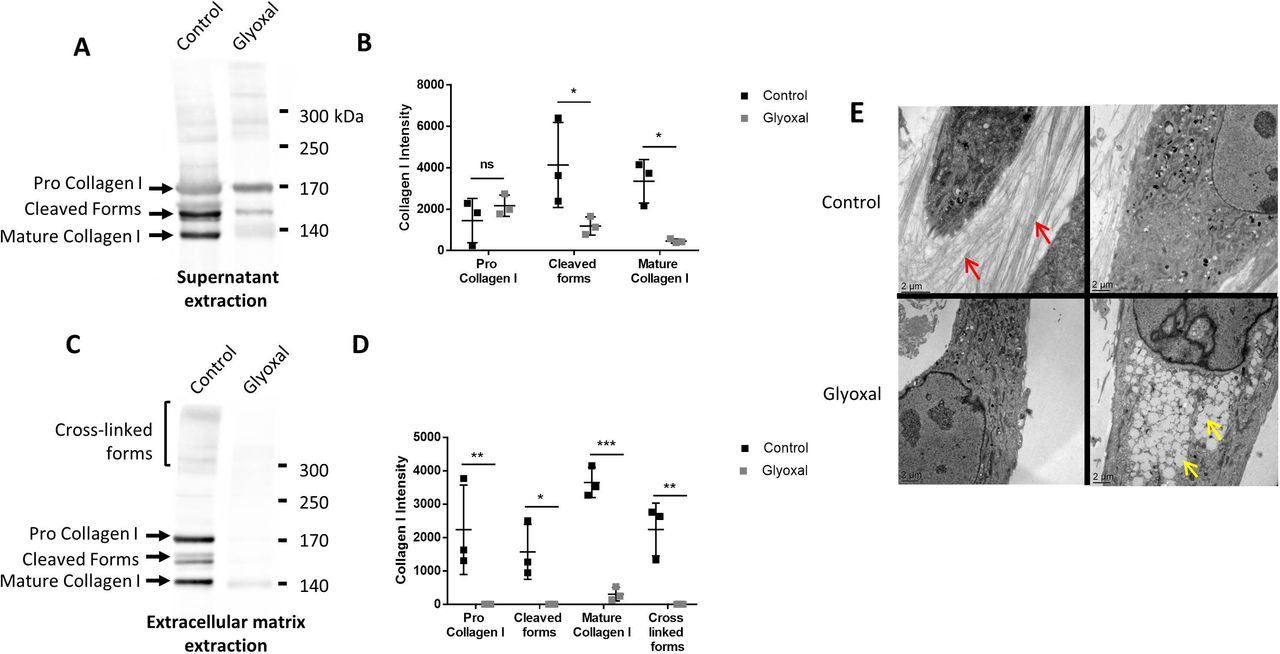
Type I collagen secretion and extracellular matrix deposition are reduced in the presence of glyoxal. The assays were performed on fibroblasts from three different donors. Control: untreated cells; glyoxal: 0.6 mM. (A) Representative Western blot analysis of secreted type I collagen. For each condition, 10 µg of protein was loaded. (B) Quantification of secreted type I collagen. (C) Representative Western blot analysis of type I collagen deposited in the extracellular matrix. (D) Quantification of collagen I deposited in the extracellular matrix. (E) Analysis of the fibroblast ultrastructure and extracellular matrix by transmission electronic microscopy. The red arrows indicate collagen fibers. The yellow arrows indicate lipid droplets. The scale bar is indicated in each image. Two-way ANOVA of the control vs glyoxal treatment is indicated as *p<0.05; **p<0.01; ***p<0.001; ns=non-significant. The data represent means±SD. ANOVA, analysis of variance.
Transmission electron microscopy allowed us to visualize the organization of the collagen fibers in the ECM. The untreated cells were able to produce numerous spatially organized collagen fibers with characteristic striations. In contrast, the glyoxal treatment resulted in no deposition of collagen fibers in the extracellular environment (figure 3E). Altogether, these results demonstrated that the glyoxal treatment did not affect collagen I secretion but it did appear to block its cleavage, maturation, and/or deposition.
Since the accumulation of lipid vesicles is implicated in the cellular and functional alteration of non-fat tissues in patients with diabetes, we examined their presence in dermal fibroblasts in response to glyoxal treatment. We found that there were empty white vesicles, characteristic of LDs, in the cytosol (yellow arrow, figure 3E). Staining with BODIPY 493/503 allowed us to determine that these white vesicles were indeed LDs (online supplemental figure S2A). On average, the diameter of the LDs was 660 nm in presence of glyoxal and ±480 nm in its absence (online supplemental figure S2B). LDs were observed in both conditions. Nevertheless, a strong accumulation of these vesicles was noted in the cytosol as a result of glyoxal treatment (figure 3E).
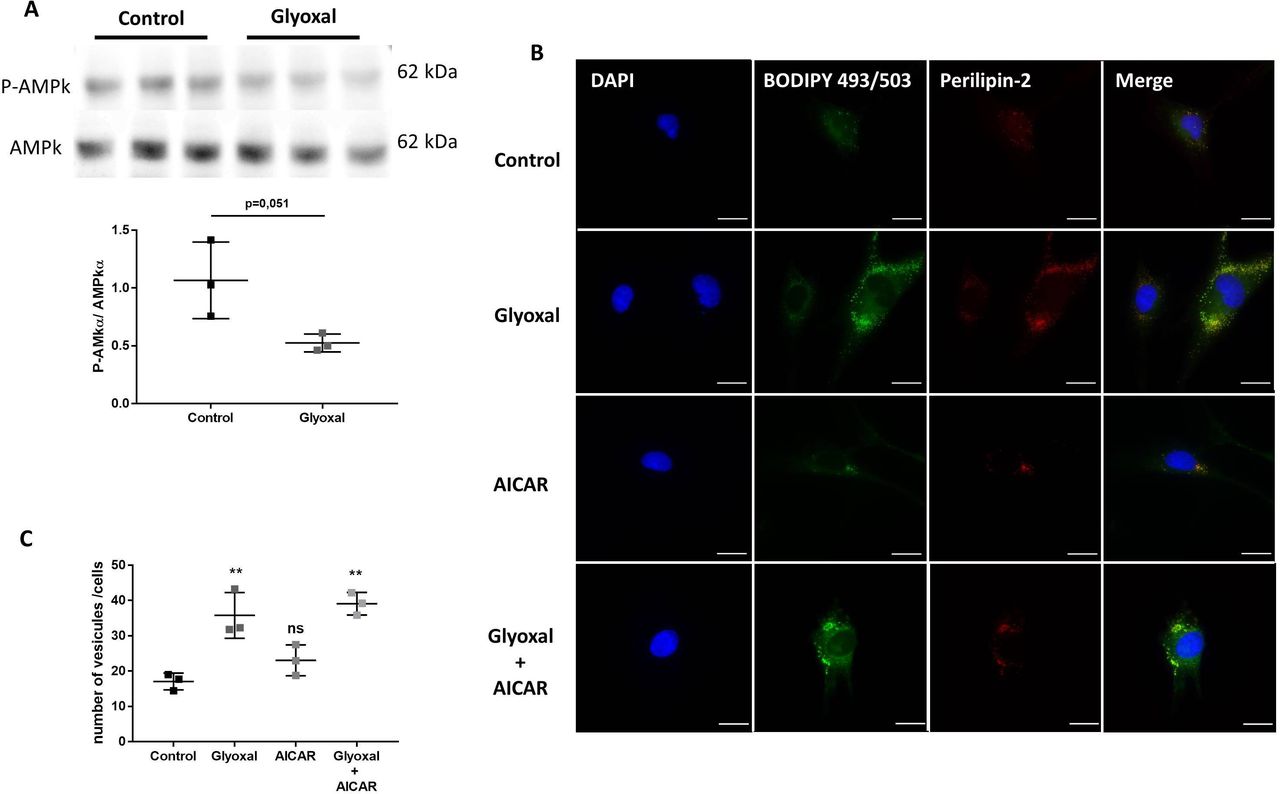
Glyoxal treatment led to accumulation of lipid vesicles and PLIN2 independently of AMPK phosphorylation
The expression of AMPKα and its phosphorylation on Thr172 can be used as a sensor of cellular energy and lipid metabolism. By Western blotting, we found that 24 hours of glyoxal treatment reduced the P-AMPK/AMPKα ratio (figure 4A).

Glyoxal treatment leads to lipid vesicle accumulation independently of AMPK. The assays were performed on fibroblasts from three different donors. Control: untreated cells; glyoxal: 0.6 mM; AICAR: 1 mM; glyoxal+AICAR: 0.6 mM glyoxal+1 mM AICAR. (A) Western blot analysis of AMPKα and P-AMPKα and measurement of the P-AMPKα/AMPKα ratio. (B) BODIPY 493/503 fluorescence in green, anti-PLIN2 staining in red, and nuclear DAPI staining in blue (scale bare=20 µm). (C) The mean number of lipid vesicles per cell for each fibroblast donor. The LDs were counted randomly (≥50 cells were counted per condition). A t-test is indicated in (A). One-way ANOVA is indicated in (C). **p<0.01; ns=non-significant. The data represent means±SD. AICAR, 5-aminoimidazole-4-carboxamideribonucleotide; ANOVA, analysis of variance; DAPI, 4',6-diaminido-2-phenylindole; LD, lipid droplet.
To determine whether exposure to glyoxal could rapidly lead to enhanced LD formation, we used BODIPY staining to determine the number of LDs. Without glyoxal treatment, there were very few LDs present in the cytosol of cells and we found that these LDs colocalized with PLIN2 (figure 4B). It appeared that 24 hours of glyoxal treatment was enough to induce extensive accumulation of LDs. This was associated with an increase in PLIN2, which appeared to fully colocalize with the LDs (figure 4B). On average, 17 vesicles per fibroblast were present without any treatment but the average number of LDs per cell reached 36 after 24 hours of glyoxal treatment (figure 4C). To verify whether the LD accumulation was dependent on AMPKα, we used AICAR to induce its phosphorylation. AICAR was able to increase the level of P-AMPK in the presence or absence of glyoxal (online supplemental figure S3). Similar to the untreated cells, there was an average of 23 LDs per cell after 24 hours of AICAR treatment (figure 4B,C). On the other hand, following glyoxal treatment, there was an average of 39 LDs per cell. Thus, AICAR did not appear to block the accumulation of LDs induced by glyoxal treatment (figure 4B,C). In the same way, PLIN2 was associated with this accumulation (figure 4B). All of these results show that glyoxal decreased AMPK phosphorylation, although this was not responsible for the lipid accumulation in cytosol of the fibroblasts.
Discussion
The present study sheds light on the effect of glycation on dermal fibroblast behavior in wound healing context. Glyoxal exposure reduced fibroblast proliferation and migration, altered the mechanostructural organization of cells, and blocked collagen deposition. These results are evidence of the pronounced effect of glycation on wound healing and skin quality (figure 5). For the first time, we show that the accumulation of LDs in dermal fibroblasts is independent of AMPK phosphorylation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
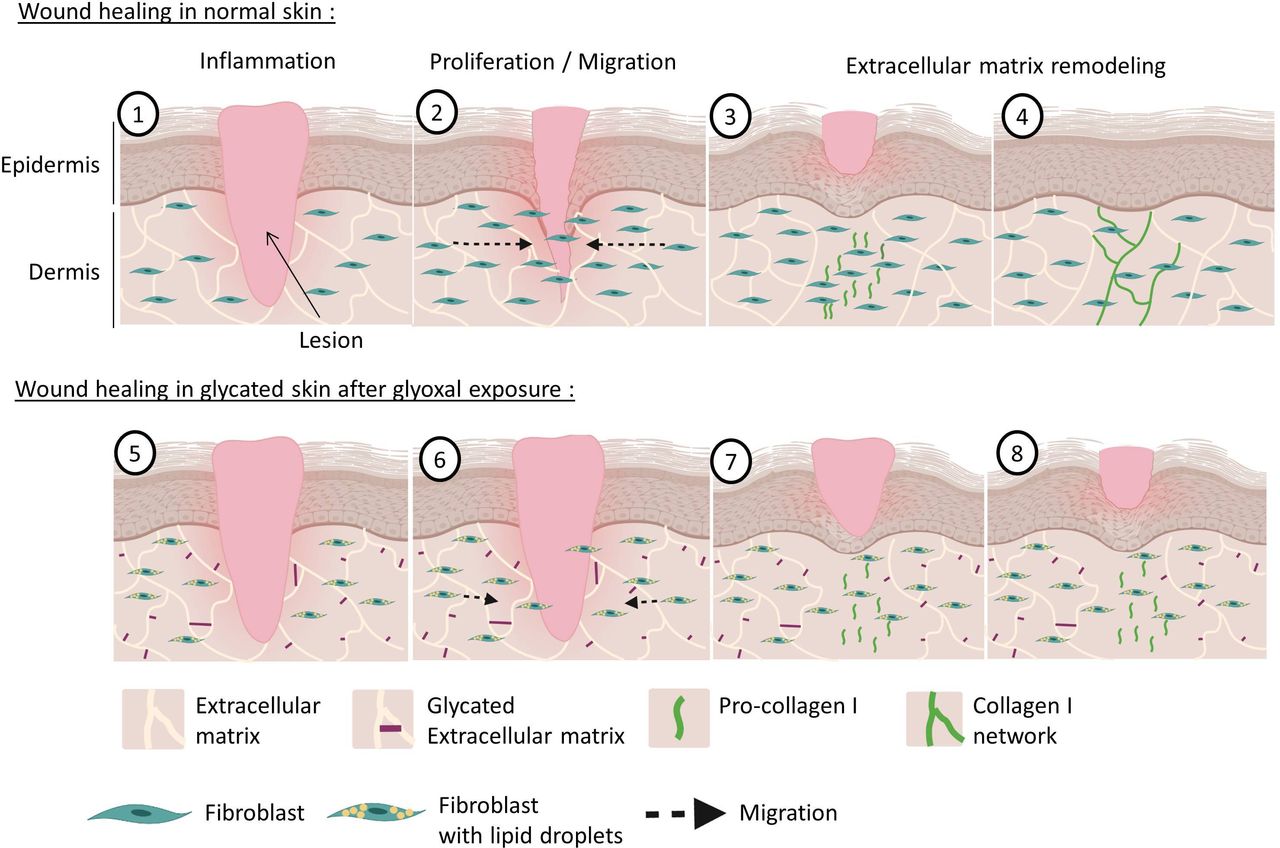
Wound healing of normal and glycated skin. During dermal wound healing following a lesion (1), fibroblasts multiply and migrate towards the center of the wound to participate in the wound closure during the proliferation phase (2). Thereafter, during the remodeling phase, the fibroblasts produce pro-collagen I (3), which matures and is deposited as a cross-linked form in the dermis to restore the skin (4). With hyperglycemia and increased glycation in diabetics, the dermal extracellular matrix becomes highly glycated (5). Our model shows that exposure to glyoxal leads to the accumulation of lipid droplets in fibroblasts, with impaired proliferative and migratory capacities (6). During the remodeling phase, pro-collagen I is produced correctly (7) but it does not mature properly and it cannot induce the formation of new collagen fibers (8).
Glyoxal is one of the highly reactive dicarbonyl species formed during glycation. Most studies use methylglyoxal or 3-DG to analyze the in vitro formation of AGEs.24 25 But glyoxal is known to produce CML,6 which is found in large quantities in the urine and serum of patients with diabetes.3 26 27 As CML is a good biomarker of diabetes, glyoxal is one of the best glycating agents for in vitro studies of glycation. We found that 0.6 mM of glyoxal induced CML production after 24 hours of treatment and that it did not induce cell death. The level of glyoxal and methylglyoxal increases in mitochondria under hyperglycemic conditions, affecting mitochondrial respiration alteration.28–30 Seahorse technology allowed us to undertake real-time analysis of cell function, and we demonstrated that the fibroblasts were metabolically active after 24 hours of glyoxal treatment. Exposure to glyoxal did not appear to result in altered mitochondrial respiration, as occurs in diabetics.28–30 It has been shown that glyoxal levels of less than 1 mM do not alter fibroblast morphology and they do not induce apoptosis,31 which is in accordance with our results.
Chronic ulcers in patients with diabetes can be due to defective fibroblast functions, such as proliferation and migration as a result of the accumulation of AGEs, and we have shown that glyoxal altered both of these behaviors. Our study examined the characteristics of fibroblasts from diabetic feet,32 and our results are concordant with those of Yoshihara et al who reported a decrease in proliferation between 3 and 5 days in culture when fibroblasts were treated with similar glycated products.33 Furthermore, using AFM, we showed that the tensile strength of the fibroblasts was greatly increased in the presence of glyoxal, thus indicating a modification of the cell mechanical properties. The fibroblasts appeared to adhere more to the ECM due to glycation, which may have reduced their ability to migrate during wound healing. The literature indicates that fibroblasts cultured on 3-DG-treated collagen were less effective at wound closure and failed to extend filipodia during migration.25 Moreover, the expression of focal adhesion kinase, which is a protein involved in cell migration, was downregulated in fibroblasts cultured on glycated collagen, thus supporting our results.25
The ECM plays an essential role in efficient wound healing and also in tissue remodeling. Collagens are the most abundant proteins in the dermis and give rise to the resistance capacities of tissues. In vitro, fibroblasts start to produce ECM when they reach confluency, and the formation of ECM readily be observed 8 days postconfluency. We hence decided to treat confluent fibroblasts with glyoxal to analyze the consequences on ECM production, maturation, and organization, as well as the effects of the presence of ascorbic acid, which allows proline hydroxylation and collagen triple helix formation. We found that under these conditions, glyoxal did not influence collagen I secretion but it appeared to reduce its maturation. Glyoxal also appeared to greatly reduce collagen deposition because no collagen fibers were present in the extracellular environment, unlike in the untreated cells. The data in the literature regarding collagen modulation according to the cell type and the induction of glycation24 34 35 are not consistent. Serban et al34 demonstrated upregulation of type I and type III collagen at the protein and gene levels after exposure of fibroblasts to AGEs-BSA. By contrast, another study found inhibition of human vaginal fibroblast proliferation in the presence of AGEs associated with a decrease in the collagen I protein level,35 which is in keeping with our results.
Transmission electronic microscopy allowed us to determine that there was pronounced accumulation of LDs in the cytosol of fibroblasts treated with glyoxal compared with control cells. Cellular lipid homeostasis is driven by LDs found in all types of mammalian cells.36 LDs are comprised of a monolayer of phospholipids that stores triacylglycerol and sterol ester. These vesicles neutralize the toxic effects of free fatty acids by converting them into neutral lipids. Fibroblasts can accumulate LDs that are 0.1–5 µm in size, and primary adipocyte LDs can reach more than 100 µm in size.37 Glyoxal affect lipid metabolism by inducing a pronounced and rapid LDs accumulation in 24 hours. We demonstrated that the accumulation of LDs by glyoxal treatment was associated with an increase in PLIN2. The expression of GFP-PLIN2 enhances LD levels in fibroblasts independently of adipocytes and lipogenic specific genes.38 PLIN2-null mice counteract insulin resistance and diet-induced obesity by increasing physical activity and by modulating food intake.20 21 Moreover, long-term treatment of 3T3-L1 fibroblasts with CML has been reported to result in extensive accumulation of LDs and to induce increased expression of miR-103 and miR-143, which are implicated in lipid accumulation.39 Yuan et al found that Oil Red O staining was enhanced in HK-2 cells in the presence of CML.40 All of these studies are fully concordant with our results. Our cellular model perfectly reproduces ectopic fat accumulation in cells from non-adipose tissue, which is a distinctive feature in metabolic damage such as diabetes.18
There have been limited studies to date regarding the link between the accumulation of LDs and impaired migration or proliferation. Mouse 3T3 fibroblasts have been shown to contain more LDs when they proliferate compared with when they are quiescent,41 which is inconsistent with our results. But Cao et al showed that renal carcinoma cells exhibited enhanced migration and invasion on PLIN-2 knockdown.42 Moreover, in injured liver hepatic stellate cells, depletion of LDs led to a higher proliferative activity and they transformed into myofibroblast-like cells to repair the tissue.43 Thus, PLIN2 appears to be a very good therapeutic target for enhancement of migratory and proliferative capacities.
Numerous studies have shown that AMPK activity is central to the formation and turnover of lipid vesicles. Our results indicate that lipid accumulation in dermal fibroblasts is independent of AMPK phosphorylation. Phosphorylation of AMPK has been shown to be required for the formation of LDs by oleic acid degradation following PLIN2 activation.44 Chen et al showed that adipogenesis can be regulated through AMPK-dependent or independent mechanisms.45 For the first time, we show that glycation can induce ectopic lipid accumulation by increasing PLIN2 in dermal fibroblasts, independently of the AMPK pathway.
Glycation is one of the main factors involved in impaired wound healing in patients with diabetes. Taken together, our results highlight that glyoxal promotes a change in fibroblast phenotype in favor of lipogenic activity that could be involved in delaying wound healing. In the context of wound healing, our study shows that glycation by glyoxal could act at the different phases of wound healing of the dermis (figure 5). The proliferative and migratory capacities of fibroblasts that accumulate lipid vesicles are altered, which may slow down wound closure and participate in the establishment of chronic wounds. In the ECM remodeling phase, glycated fibroblasts would be able to produce pro-collagen I, but cannot then mature normally to induce the formation of a new collagen I network. These observations could to a certain extent explain the skin fragility observed in patients with diabetes.
In vitro, glyoxal treatment of fibroblasts could be an excellent cell model for studying skin complications related to diabetes and it could help with testing new therapeutic approaches to improve wound healing. It would be interesting to select therapeutic molecules that may enhance key properties of fibroblasts such as proliferation, migration, collagen production, and lipid metabolism.
Data availability statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
Acknowledgments
The flow cytometry experiments were performed with the help Remy Pépin at the LOEX (Research center of University Hospital Center of Québec—Laval University, Québec City, Canada). We thank Christophe Chamot at the PLATIM (SFR Biosciences, Gerland, Lyon, France) for his technical assistance with the time-lapse experiments. We are grateful to the BioMeca SAS (Lyon, France) and particularly to Julien Chlasta and Anna Catte-Drillat for performing the AFM experiments and for their excellent analysis and advice. The TEM experiments were performed by Christelle Fabrer-Boule at the CTµ (Centre Technologique des Microstructures, Villeurbanne, France). Last, we also thank Laurence Canaple at the Plateau de Recherche Expérimentale en Criblage In Vivo (SFR Biosciences, Lyon, France) for her excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DS-R and CB are joint senior authors.
Presented at Parts of this study were presented at the ‘7th Skin Physiology International Meeting’ in France in 2020 and at ‘Club de Recherches Cliniques du Québec’ in Estérel, Canada in 2019.
Contributors DS-R, MB, SC, CB, and CG designed the research project. CG, CB, and SF performed the experiments. CG, CB, SF, MB, SC, and DS-R contributed to analysis and interpretation of the data. CG and CB wrote the paper, and DS-R, MB, SC, and SF edited the manuscript.
Funding Urgo Research Innovation and Development (URID, Chenôve, France) supported the study.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.