Article Text

Abstract
Introduction Diabetic ketoacidosis (DKA) causes acute and chronic neuroinflammation that may contribute to cognitive decline in patients with type 1 diabetes. We evaluated the effects of agents that reduce neuroinflammation (triarylmethane-34 (TRAM-34) and minocycline) during and after DKA in a rat model.
Research design and methods Juvenile rats with DKA were treated with insulin and saline, either alone or in combination with TRAM-34 (40 mg/kg intraperitoneally twice daily for 3 days, then daily for 4 days) or minocycline (45 mg/kg intraperitoneally daily for 7 days). We compared cytokine and chemokine concentrations in brain tissue lysates during DKA among the three treatment groups and in normal controls and diabetic controls (n=9–15/group). We also compared brain inflammatory mediator levels in these same groups in adult diabetic rats that were treated for DKA as juveniles.
Results Brain tissue concentrations of chemokine (C-C) motif ligand (CCL)3, CCL5 and interferon (IFNγ) were increased during acute DKA, as were brain cytokine composite scores. Both treatments reduced brain inflammatory mediator levels during acute DKA. TRAM-34 predominantly reduced chemokine concentrations (chemokine (C-X-C) motif ligand (CXCL-1), CCL5) whereas minocycline had broader effects, (reducing CXCL-1, tumor necrosis factor (TNFα), IFNγ, interleukin (IL) 2, IL-10 and IL-17A). Brain inflammatory mediator levels were elevated in adult rats that had DKA as juveniles, compared with adult diabetic rats without previous DKA, however, neither TRAM-34 nor minocycline treatment reduced these levels.
Conclusions These data demonstrate that both TRAM-34 and minocycline reduce acute neuroinflammation during DKA, however, treatment with these agents for 1 week after DKA does not reduce long-term neuroinflammation.
- Diabetic Ketoacidosis
- Brain
- Cytokines
- Inflammation
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
Previous studies in both rodent models and humans suggest that diabetic ketoacidosis (DKA) causes acute neuroinflammation that may result in brain injury. Studies in rodent models also suggest that DKA might trigger chronic neuroinflammation that could contribute to diabetes-related cognitive decline. Whether pharmaceutical interventions can be used to reduce neuroinflammation resulting from DKA is unknown.
What this study adds
The current study demonstrates that administration of either TRAM-34 or minocycline during DKA treatment with insulin and saline in a rat model reduces acute neuroinflammation, however, treatment with either of these agents for 1 week after recovery from DKA did not reduce long-term neuroinflammation in adult rats that had DKA as juveniles.
How this study might affect research, practice and/or policy
Treatment with pharmacological agents that reduce neuroinflammation should be studied further as a means of reducing acute brain injury caused by DKA in children. Whether chronic neuroinflammation resulting from DKA can be reduced by antineuroinflammatory agents remains unclear and will require evaluation of longer treatment durations after DKA recovery.
Introduction
Diabetic ketoacidosis (DKA) occurs in 30%–60% of children with new onset of type 1 diabetes (T1D) and may occur subsequently (7%–10% per year) during episodes of illness, malfunction of diabetes care equipment or diabetes mismanagement.1–4 Recent studies have linked DKA to declines in cognition in children with T1D, demonstrating associations between DKA and reductions in IQ and memory function, as well as structural alterations in the brain.5–10
In previous work by our group using a juvenile rat model, we found that DKA causes a neuroinflammatory response with reactive astrogliosis and activation of microglia,11–13 along with increased levels of inflammatory mediators (cytokines, chemokines and matrix metalloproteinases (MMPs)) in brain tissue lysates.14 Notably, we found evidence that neuroinflammation persists into adulthood in rats exposed to DKA as juveniles,14 suggesting that DKA might trigger chronic low-level neuroinflammation. In the current study, we investigated the effects of pharmacological agents that reduce neuroinflammation (triarylmethane-34 (TRAM-34) and minocycline) on acute and long-term neuroinflammation and systemic inflammation in a rat DKA model.
Methods
This study was conducted in accordance with the Animal Use and Care Guidelines issued by the National Institutes of Health.
Overview
In these studies, we determined the effects of TRAM-34 and minocycline on levels of inflammatory mediators in the blood and brain of juvenile rats during acute DKA and adult rats exposed to DKA as juveniles. In the first series of experiments, we generated DKA in juvenile rats and treated DKA with either insulin and saline alone or insulin and saline plus TRAM-34 or minocycline. We measured levels of cytokines and chemokines in blood samples and brain tissue lysates in each group of rats during DKA treatment, as well as normoglycemic (NG) and hyperglycemic (HG) control rats. In the second series of experiments, rats in each group were treated for DKA with either insulin and saline alone or insulin and saline plus TRAM-34 or minocycline. After recovery from DKA, rats received insulin treatment until adulthood (70 days of age). We measured levels of cytokines and chemokines in blood samples and brain tissue lysates of these adult rats as well as adult rats of the same age and diabetes duration without past exposure to DKA.
Juvenile rat diabetes model
Four weeks to 5 weeks old Sprague Dawley rats (n=9–15 per group, 100 g, male and female, Charles River Laboratories, Wilmington, Massachusetts, USA) were treated with an intraperitoneal (IP) injection of streptozotocin (STZ, Sigma-Aldrich, St. Louis, Missouri, USA) 165 mg/kg to induce diabetes. NG control rats received an IP injection of STZ vehicle (citric acid, Sigma-Aldrich, St. Louis, Missouri, USA). The rats’ drinking water was replaced with water containing 10% dextrose (Fisher Scientific, Santa Clara, California, USA) for 24 hours after STZ treatment to prevent hypoglycemia. Urine glucose and acetoacetate were measured daily (Multistix urinalysis strips, BAYER, Fisher Scientific, Santa Clara, California, USA). Beginning 24 hours after STZ injection, rats were treated with subcutaneous (SC) insulin (Novolin 70:30 insulin (Novo Nordisk, Princeton, New Jersey, USA), 1 unit in the morning and 2 units in the evening). STZ-treated rats in the HG control group continued to receive SC insulin injections twice daily for the duration of the experiments. In the DKA groups, SC insulin injections were given for 6 days after STZ, after which they were withheld to induce DKA.
DKA generation and treatment
In the DKA groups, insulin was withheld for a 24-hour period during which time rats were given a high fat diet (diet D12492, 60 kcal% fat, Research Diets, New Brunswick, New Jersey, USA). Water was removed for 12 hours to promote ketosis and increase dehydration. When urine glucose rose above 2000 mg/dL and urine acetoacetate above 160 mg/dL, blood analyses were conducted to confirm DKA (blood glucose ≥300 mg/dL and β-hydroxybutyrate >3 mmol/L) using Precision Xtra glucose and ketone monitoring system (Abbott Diabetes Care, Alameda, California, USA).
To treat DKA, rats initially received an IP injection of 0.9% saline (8 mL/100 g body weight) and SC regular human insulin (Novolin R, Novo Nordisk, Princeton, New Jersey, USA, 0.5 units/100 g body weight). Subsequently, rats received 0.9% saline (4 mL/100 g body weight IP) and regular human insulin (0.5 units/100 g body weight SC) hourly. Blood glucose and β-hydroxybutyrate were measured every 2 hours during treatment. Serum electrolytes and pH were measured before and after treatment (I-STAT Portable Clinical Analyzer; Sensor Devices, Waukesha, Wisconsin, USA). In experiments involving full recovery from DKA (blood glucose concentrations below 200 mg/dL, venous pH above 7.30 and serum β-hydroxybutyrate concentrations below 0.5 mmol/L), insulin and saline treatment times varied between 5 hours and 8 hours. When DKA was resolved, rats received an SC injection of 2 units of Novolin 70:30 insulin, and then resumed twice daily insulin injections as prior to DKA induction. NG and HG control groups underwent identical sham treatments (IP puncture, SC puncture, blood sampling).
TRAM-34 and minocycline treatment
Rats with DKA were randomized to either standard DKA treatment (insulin and saline as described above) alone, standard DKA treatment plus TRAM-34 or standard DKA treatment plus minocycline. Rats in the TRAM-34 group received an IP injection of TRAM-34 (40 mg/kg body weight)15–17 30 min before beginning insulin/saline treatment, then every 12 hours for 3 days, then daily for four additional days. TRAM-34 was dissolved in medium chain triglycerides (Miglyol-812 caprylic/capric triglyceride, Spectrum Chemicals) and injected in a volume of 2µL/g body weight. Miglyol-812 is a low viscosity oily vehicle which is widely used as an excipient and well tolerated for IP, SC or oral administration. The minocycline group received an IP injection of minocycline (45 mg/kg body weight, TCI America, Portand, Oregon, USA)18–20 administered at the beginning of insulin/saline treatment and daily thereafter for 7 days. Minocycline was dissolved in phosphate-buffered saline and injected in a volume of 0.9µL/g body weight. Rats in the standard DKA treatment (control) group received IP injections of either TRAM-34 vehicle (Miglyol-812) or minocycline vehicle (phosphate-buffered saline), with half of the group receiving TRAM-34 vehicle and the other half minocycline vehicle.
Blood and brain sample collection
In the first series of experiments, blood and brain tissue samples were obtained during acute DKA, 4 hours after beginning insulin and saline treatment (approximating the peak time for symptomatic brain injury in children with DKA). In the second series of experiments, blood and brain tissue samples were obtained when the rats reached adulthood (70 days of age, 28 days after DKA resolution). Blood and brain tissue samples were obtained from HG control rats of the same age and diabetes duration as the acute DKA group and the postrecovery group. NG control blood and brain tissue samples were collected from rats identical in age to the acute DKA group.
Rats were anesthetized using isoflurane and blood samples were collected from the left femoral artery. After blood samples were obtained, rats were decapitated using a guillotine. Brain samples were collected and dissected into regions according to previously published methods.21 The left hemisphere was harvested intact. The right hemisphere was dissected into regions including cortex and hippocampus. All brain samples were typically collected within 10–12 min. Brain samples were initially frozen in microcentrifuge tubes placed in dry ice (−20°C) and then stored at −80°C.
Blood sample processing and brain lysate preparation
Blood samples were allowed to clot for 10 min, then centrifuged at 12 000 rpm for 5 min at room temperature. Serum (100–200 µL) was collected and stored at −80°C. Brain tissue lysate processing followed previously published methods.22 Brain samples were homogenized with tissue protein extraction reagent (TPER, Thermo Fisher Scientific, Waltham, Massachusetts, USA) and Halt Protease Inhibitor Cocktails (Thermo Fisher Scientific, Waltham, Massachusetts, USA). 2 mL of TPER was used for every 100 mg of brain tissue with 1 uL of protease inhibitor added for every 100 uL of TPER. Tissue was homogenized with a Dounce homogenizer and then processed with an ultrasonicator (Branson Sonifier 4C15, three pulses of 1 s each at 4°C). Lysate supernatant was collected after samples were centrifuged at 16 000 rpm for 10 min at 4°C.
Cytokine and chemokine assays
We used multiplex solid phase sandwich immunoassays (Invitrogen ProcartaPlex custom plates, Thermo Fisher Scientific, Waltham, Massachusetts, USA) to measure cytokine and chemokine concentrations (chemokine (C-X-C) motif ligand (CXCL1), chemokine (C-C) motif ligand (CCL)3, CCL5, tumor necrosis factor (TNFα), interferon (IFNγ), interleukin (IL) 2, IL-10, IL12p70 and IL-17A) in serum and brain lysates. Cytokines and chemokines were chosen based on previous studies identifying inflammatory mediators that were altered by DKA in brain tissue lysates.14 Microbeads of defined spectral properties coated with analyte-specific antibodies were incubated with brain tissue lysate or serum to capture the analytes. After washing, the beads were reacted with analyte-specific biotinylated detector antibodies which bound to the appropriate immobilized analytes. After additional washing, the beads were reacted with streptavidin-R-phycoerythrin (RPE) which bound to the biotinylated detector antibodies associated with the immune complexes on the beads, forming a four-member solid phase sandwich. After final washing to remove unbound material, the beads were analyzed in a Luminex instrument. The concentration of each analyte was determined by monitoring the spectral properties of the beads and amount of associated RPE fluorescence for each sample and comparing to standards.
Statistical analyses
Statistical analysis was performed in Stata/SE V.16. To promote comparability across analytes, support creation of composite variables, and minimize the influence of skewed or outlying values, all cytokine and chemokine measurements were natural log transformed and then robustly z-score transformed. For each analyte and study phase (acute vs day-28), the robust z-score was computed in three steps. First, batch-specific medians from an appropriate comparison group were computed and then subtracted from each measurement to center them. For acute and day-28 measurements, the comparison groups were NG and HG, respectively. Then, a quantile (median) regression was fit to the centered values from all groups to generate residuals that could then be used to estimate a standard robust measure of spread that could be used as the divisor for standardizing the centered values, the median absolute residual divided by the 75th percentile of the standard normal distribution, approximately 0.6745. The standardized centered values were then Winsorized to lie between −3 and 3. For each animal and brain tissue location, samples with five or more inflammatory mediator measurements outside ±3 SD were considered outliers. For serum analyses, samples with three or more measurements outside ±3 SD were considered outliers. These samples were removed from the analyses (4 of 195 for acute brain, 2 of 60 from acute serum, 4 of 144 for day-28 brain, and 5 of 52 day-28 serum). We performed sensitivity analyses that included the outliers to verify similar results. Finally, for each study phase and analyte, we averaged together the animal’s brain-region specific robust z-scores to produce one analysis record per animal. To improve the precision of estimates, we also developed composite measurements based on empirical observations. For candidate items in a composite, internal consistency and item–rest correlations were conducted prior to forming the composite to verify the acceptability of combining the items together.
At each study phase and for each analyte and each composite measurement, pairwise contrasts among group means were estimated using the xtmixed command for general linear models with robust variance estimates to protect against unbalanced group sizes, heteroskedasticity and slight departures of measurement errors from normality assumptions. Electrolyte, glucose and pH values were compared among groups using one-way analysis of variance with Bonferroni post hoc testing (Stata/SE V.11).
Results
Glucose and electrolyte concentrations and pH values for rats in each of the experimental groups before beginning DKA treatment are presented in tables 1 and 2. Similar biochemical values were observed in all DKA groups before treatment.
Acute DKA experiments. Biochemical values in each comparison group at the time of initiation of DKA treatment, mean (SD)
Long-term experiments. Biochemical values in each comparison group at the time of initiation of DKA treatment in the juvenile period, mean (SD)
Acute DKA
To determine the effects of TRAM-34 and minocycline on neuroinflammation and systemic inflammation during acute DKA, we compared levels of inflammatory mediators in rat brain and serum specimens during acute DKA (after 4 hours of treatment with insulin and saline, coinciding with the peak time of cerebral injury in children with DKA).
Serum
Similar to our previous work,14 we found that acute DKA was characterized by elevations in serum concentrations of CCL3, TNFα, IL-2 and IL-10 (figure 1). Cytokine composite scores (mean Z-scores for highly correlated serum inflammatory mediators including CXCL1, CCL3, CCL5, TNFα, IL-2, IL-10 and IL-17A, Cronbach’s α=0.73) were also significantly elevated during DKA. Treatment with TRAM-34 during acute DKA reduced serum cytokine composite scores, however, minocycline had no significant effect on cytokine composite scores. Effects of the anti-inflammatory interventions on concentrations of individual inflammatory mediators also differed. Compared with standard DKA treatment, rats treated with TRAM-34 had significantly reduced serum concentrations of CXCL-1, whereas those treated with minocycline had reduced serum concentrations of CCL3 and increased concentrations of IL-17A.
Serum levels of inflammatory mediators during acute diabetic ketoacidosis (DKA). CCL, chemokine (C-C) motif ligand; CXCL-1, chemokine (C-X-C) motif ligand; DKA, DKA treated with insulin/saline; DKA-M, DKA treated with insulin/saline plus minocycline; DKA-T, DKA treated with insulin/saline plus TRAM-34; HG, hyperglycemic control; IFN, interferon; IL, interleukin; NG, normoglycemic control; TNF, tumor necrosis factor; TRAM-34, triarylmethane-34.
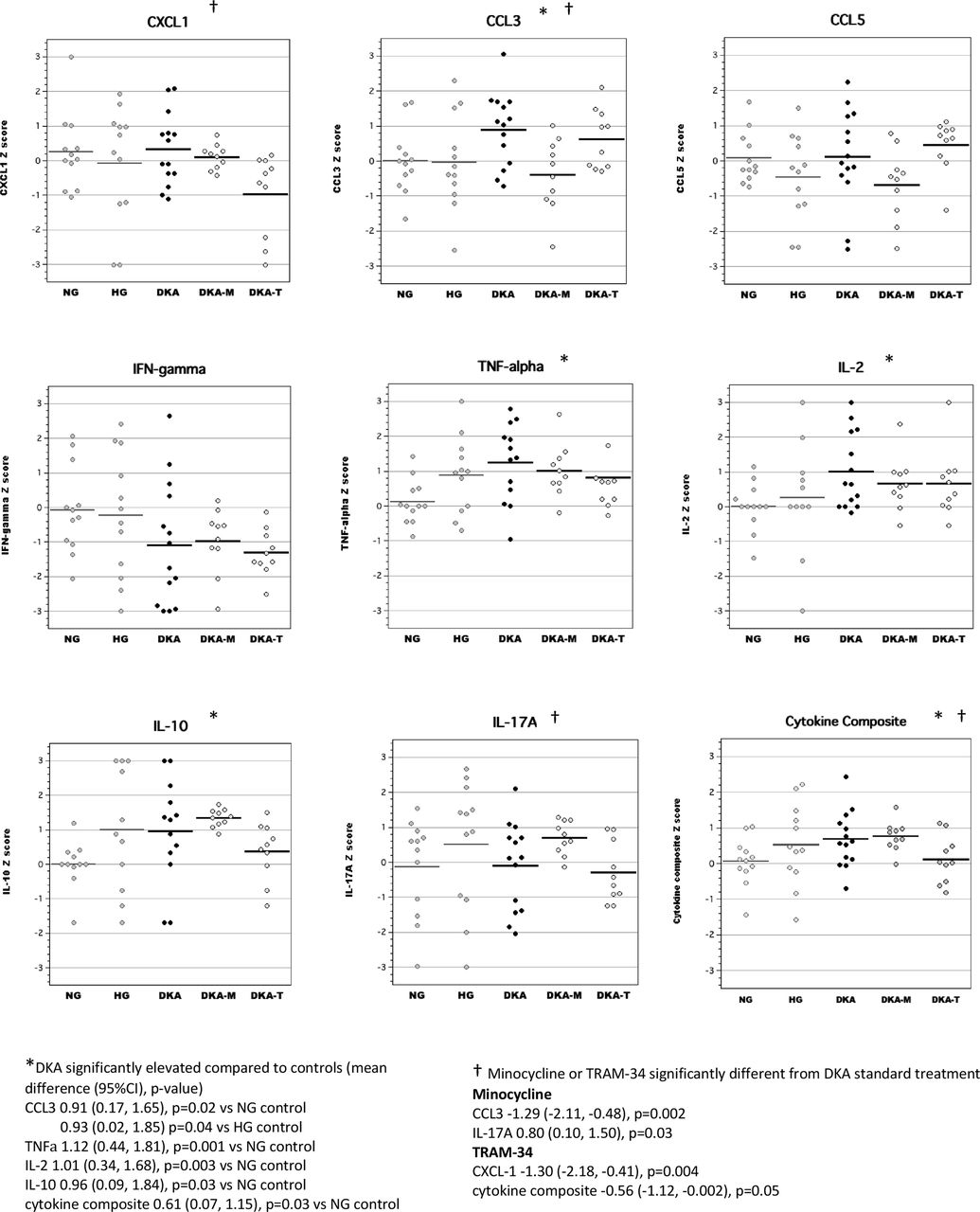
Brain tissue
In brain tissue lysates, we found that acute DKA was associated with increased levels of CCL3, CCL5 and IFNγ, similar to our previous work (figure 2).14 Cytokine composite scores (mean Z-scores for highly correlated brain inflammatory mediators including CXCL-1, CCL3, CCL5, TNFα, IFNγ, IL-2 and IL-17A, Cronbach’s α=0.84) were also significantly elevated during DKA. Both TRAM-34 and minocycline treatment during acute DKA significantly reduced brain cytokine composite scores. Minocycline broadly reduced levels of several inflammatory mediators in the brain including CXCL-1, IL-2, IL-10, TNFα, IFNγ, and IL-17A, whereas TRAM-34 had more selective effects that targeted chemokines (CXCL-1 and CCL5).

Levels of inflammatory mediators in brain tissue lysates during acute diabetic ketoacidosis (DKA). CCL, chemokine (C-C) motif ligand; CXCL-1, chemokine (C-X-C) motif ligand; DKA, DKA treated with insulin/saline; DKA-M, DKA treated with insulin/saline plus minocycline; DKA-T, DKA treated with insulin/saline plus TRAM-34; HG, hyperglycemic control; IFN, interferon; IL, interleukin; NG, normoglycemic control; TNF, tumor necrosis factor; TRAM-34, triarylmethane-34.
Long-term effects of DKA
To determine whether treatment with either TRAM-34 or minocycline could alter long-term neuroinflammation caused by DKA, we measured serum and brain cytokine and chemokine levels in adult diabetic rats (70 days old) who had experienced DKA as juveniles (28 days prior). DKA was treated either with insulin and saline alone or with the addition of minocycline or TRAM-34 during insulin/saline treatment and for 7 days after DKA recovery.
Serum
There were no significant differences in serum cytokine concentrations in adult diabetic rats with and without prior exposure to DKA (online supplemental figure 1). However, serum concentrations of several cytokines and chemokines in the minocycline group were lower than those in the HG control group, including IL-2, TNFα, IFNγ and IL17A. In contrast, CCL5 levels in the minocycline group were elevated compared with HG controls and the TRAM-34 group. Serum cytokine and chemokine levels in adult rats in the TRAM-34 group were not significantly different from those of DKA rats that received conventional DKA treatment or those of HG controls.
Supplemental material
Brain tissue
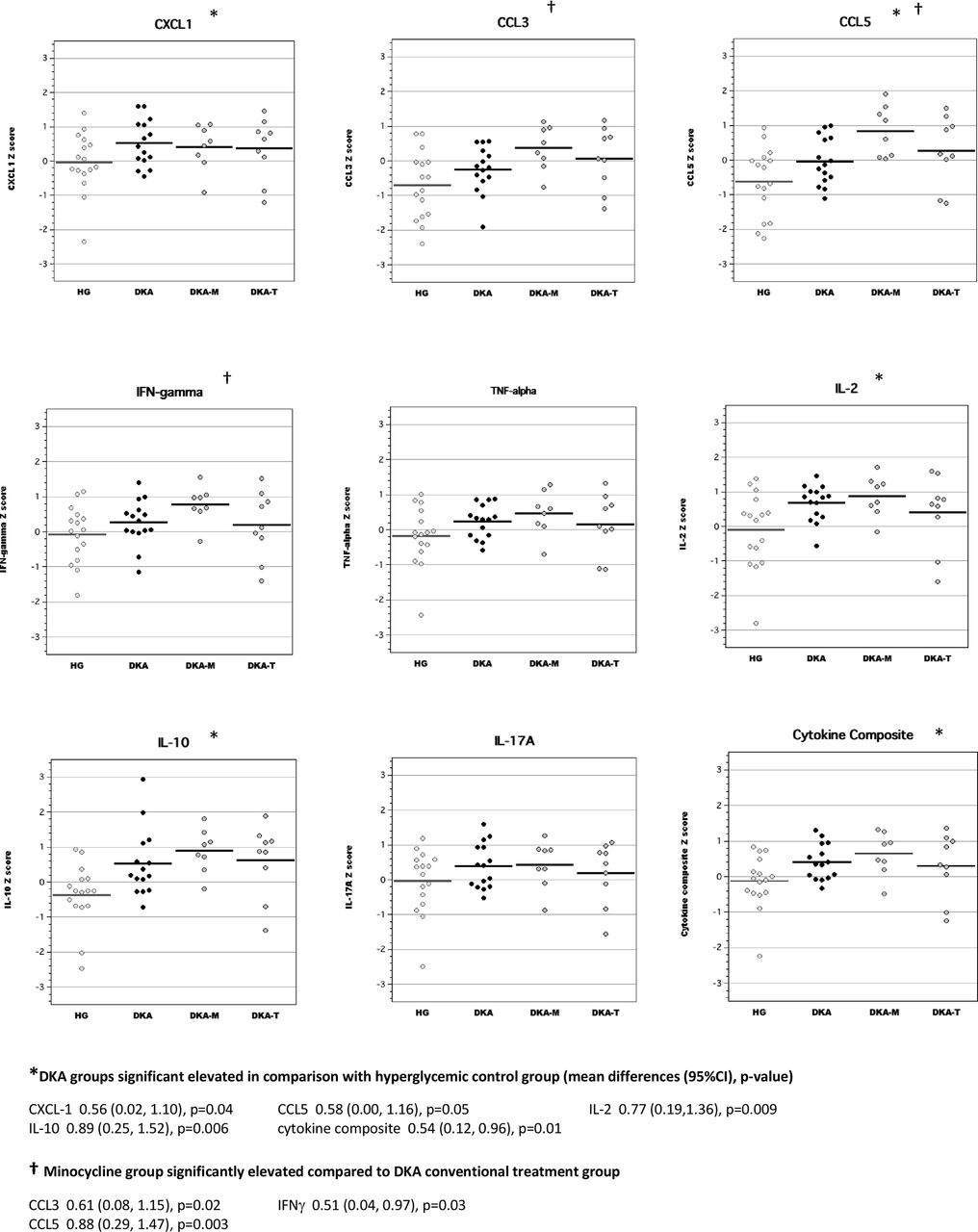
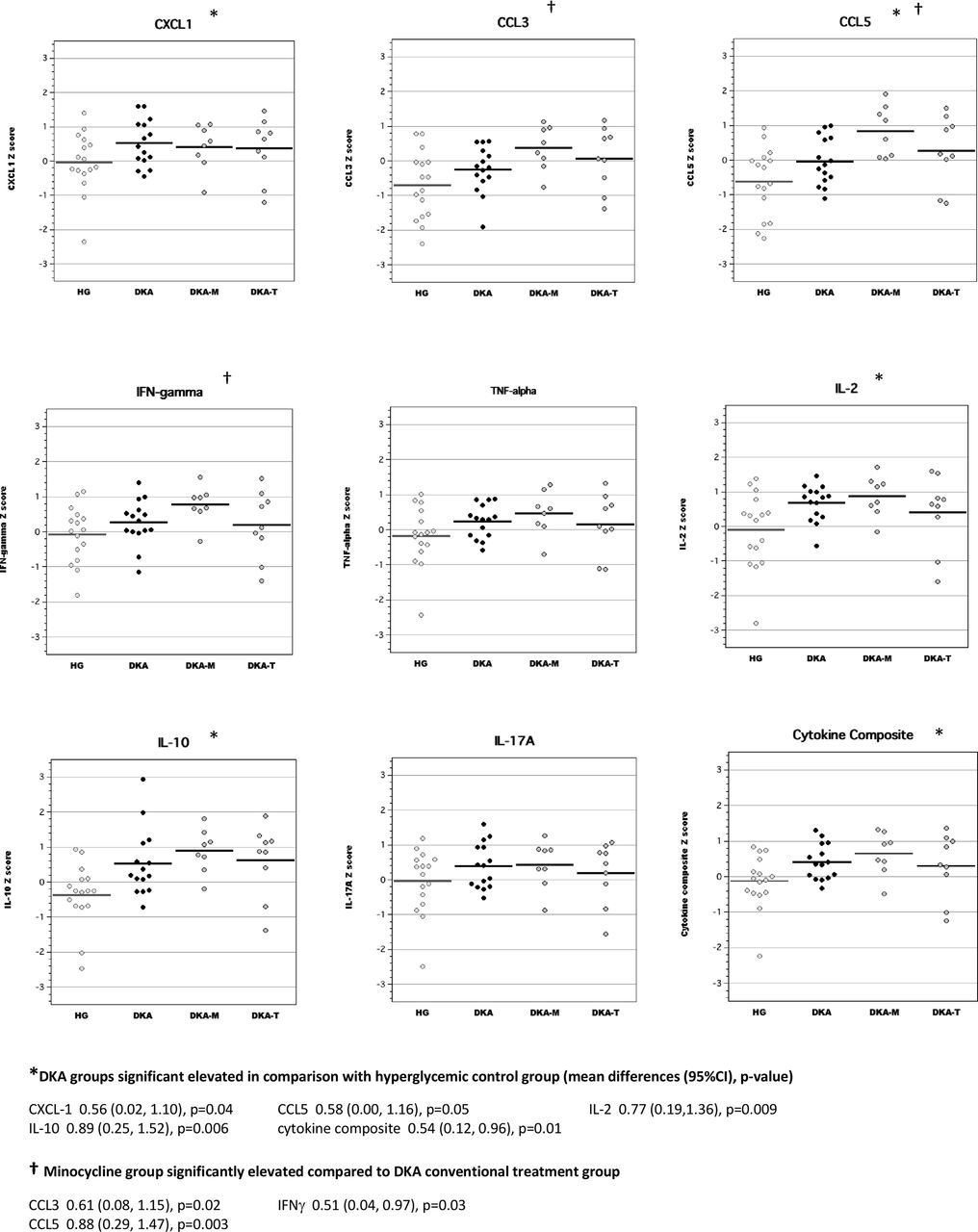
In brain tissue lysates of adult rats, exposure to DKA as juveniles resulted in elevated concentrations of CCL3, CCL5, IL-2 and IL-10 compared with adult diabetic rats without previous exposure to DKA (figure 3). Brain cytokine composite scores were also higher in adult diabetic rats with previous DKA exposure. Interestingly, brain levels of IL-2 and IFNγ in the minocycline group were significantly higher than those of the conventional DKA treatment group. Brain levels of inflammatory mediators in the TRAM-34 group were not significantly different from those of rats in the conventional DKA treatment group.
{kind=link}
{kind=link}
{kind=link}
Levels of inflammatory mediators in brain tissue lysates of adult diabetic rats with and without exposure to diabetic ketoacidosis (DKA) as juveniles. CCL, chemokine (C-C) motif ligand; CXCL-1, chemokine (C-X-C) motif ligand; DKA, DKA treated with insulin/saline; DKA-M, DKA treated with insulin/saline plus minocycline; DKA-T, DKA treated with insulin/saline plus TRAM-34; HG, hyperglycemic control; IFN, interferon; IL, interleukin; TNF, tumor necrosis factor; TRAM-34, triarylmethane-34.
Discussion
Accumulating evidence suggests that DKA may cause acute brain injury in children23 and is associated with long-term decline in cognition.5 6 10 In previous studies using a rat model, we showed that acute DKA causes both systemic inflammation and neuroinflammation.14 Although systemic inflammation mainly resolved after recovery from DKA, elevated levels of cytokines and chemokines in brain tissue lysates persisted for prolonged periods and were still present in adult rats that had experienced DKA as juveniles.14 In the current study, we found that both TRAM-34 and minocycline reduced the neuroinflammatory response to acute DKA, with TRAM-34 selectively affecting chemokine levels and minocycline having broader effects. In contrast, neither treatment reduced long-term neuroinflammation resulting from DKA. This study is the first to explore the effects of neuroprotective interventions during DKA. Our data suggest potential benefits of both TRAM-34 and minocycline treatment for reducing neuroinflammation during acute DKA, but no beneficial effects on chronic DKA-related neuroinflammation. It should be noted, however, that rats were treated with neuroprotective agents for only 1 week after DKA recovery and effects of more prolonged treatment have not been explored.
The pathophysiology of DKA-related cerebral injury, both during acute DKA and over prolonged periods after DKA, is not well characterized. Recent evidence suggests that neuroinflammation plays an important role. Elevated levels of proinflammatory cytokines have been found in adolescents with fatal brain injuries resulting from acute DKA.24 25 In a mouse model, DKA increased serum levels of IL-6, IL-8, monocyte chemoattractant protein (MCP-1) (CCL2), IL-10, sE-selectin, soluble intercellular adhesion molecule (sICAM-1), and soluble vascular cell adhesion molecule (sVCAM-1).26 Stimulation of cultured brain microvascular endothelial cells with mouse DKA plasma caused cellular activation (increased reactive oxygen species (ROS) and activation of nuclear factor-kappa B (NF-κΒ)), upregulated E-selectin, ICAM-1, and VCAM-1, and increased leukocyte adhesion.26 Plasma from children with DKA, as well as mixtures of cytokines simulating those in DKA plasma, also increased leukocyte adhesion to cultured brain microvascular endothelial cells.27 Furthermore, several studies document alterations in levels of MMPs during DKA in both humans and rodents, most notably, increased levels of MMP-9.28–30 MMPs act as mediators of blood-brain barrier dysfunction in disease states characterized by systemic inflammation and brain injury.31–33 MMPs degrade tight junction proteins and endothelial basement membranes, allowing circulating proinflammatory proteins and leukocytes to invade the central nervous system.31 34 35 Release of destructive azurophilic enzymes by polymorphonuclear leukocytes during DKA has also been demonstrated and may cause blood-brain barrier degradation.36 These mechanisms provide means by which the systemic inflammatory response may contribute to brain injury.
Our current and previous studies have documented elevated levels of cytokines and chemokines in brain tissue lysates during acute DKA that persist for prolonged periods after recovery from DKA.14 The most marked elevations were observed in levels of the chemokines CXCL-1, CCL3 and CCL5. Chemokines are produced by brain microvascular endothelial cells as well as by microglia and astrocytes and are important mediators of neuroinflammation.37–43 Elevated chemokine levels promote accumulation of microglia in regions of brain injury and cause increased production of proinflammatory cytokines.37 41–43 Chemokines also attract circulating leukocytes to brain tissues resulting in brain injury via production of proteases and ROS and further damage to the blood-brain barrier.44
In previous studies, we have demonstrated activation of microglia and development of reactive astrogliosis during DKA.11 These findings are present during untreated DKA and become more pronounced during treatment with insulin and saline, suggesting that inhibition of microglial activation during DKA treatment might be an important target to diminish neuroinflammation.
TRAM-34 is a specific inhibitor of KCa3.1,45 a calcium-activated potassium channel involved in activation of microglia, the brain’s resident immune cells.16 Data suggest that inhibition of KCa3.1 may limit destructive microglial activities without affecting beneficial microglial functions.46 In rodent models, treatment with TRAM-34 reduces infarct volume after ischemic stroke,16 17 and improves pathology in a model of amyotrophic lateral sclerosis.47 48 Treatment with the structurally related KCa3.1 inhibitor, senicapoc, has beneficial effects in rodent models of Alzheimer’s disease.49 In previous studies using a rat model of DKA, we found that inhibition of microglial activation with TRAM-34 substantially reduced reactive astrogliosis in the hippocampus during DKA treatment suggesting reduced neuroinflammation and/or reduced neural injury.12 Whether reduced production of inflammatory mediators was involved in the response to TRAM-34, and which mediators were most affected, was unknown. The current study demonstrates that TRAM-34 reduces circulating levels of CXCL-1 and brain levels of CXCL-1 and CCL5 during acute DKA. TRAM-34 also reduced brain cytokine composite scores, suggesting an overall reduction in neuroinflammation. Treatment with TRAM-34 for 1 week after DKA, however, had no long-term effects on neuroinflammation resulting from DKA.
Minocycline is an antibiotic which has been found to have anti-inflammatory and neuroprotective effects. Minocycline has broader effects than TRAM-34, attenuating neuronal apoptosis and suppressing production of ROS in addition to inhibiting microglial activation.50–52 In the current studies, treatment with minocycline during acute DKA reduced brain levels of multiple inflammatory mediators and reduced brain cytokine composite scores, suggesting an overall reduction in neuroinflammation. Similar to TRAM-34, however, we did not observe improvements in long-term neuroinflammation with minocycline treatment. In fact, our long-term studies found elevated levels of some inflammatory mediators in the minocycline group, suggesting a possible rebound effect resulting in a delayed increase in neuroinflammation.
The current study has some limitations. First, a limited number of cytokines and chemokines were measured. These were chosen based on previous studies of DKA in humans and rodent models, however, it is likely that DKA, and the neuroprotective agents we evaluated, have effects on inflammatory mediators beyond those described here. In addition, our findings describe neuroinflammation and responses to treatment in a rodent model of DKA. Human neuroinflammatory responses may differ from those observed in these studies. Furthermore, several aspects of the rodent DKA model differ from human DKA. Limited water intake and a high fat diet is necessary to reliably induce DKA in rats. Increased dietary fat intake may contribute to systemic inflammation and neuroinflammation, however, lipolysis with elevated blood lipid levels is characteristic of human DKA, making the marginal contribution of dietary fat intake less significant. The vehicles used for IP administration of the pharmacological agents and the IP injections themselves may also have contributed to inflammation, however, identical procedures were used in control animals, such that between-group comparisons continue to be relevant. Most importantly, treatments were only administered for a relatively brief period (1 week) after recovery from DKA. This treatment did not reduce chronic neuroinflammation resulting from DKA, however, more prolonged treatment may have had a greater effect.
In summary, our data demonstrate that both TRAM-34 and minocycline reduce the acute neuroinflammatory response during DKA. Treatment with TRAM-34 mainly affected chemokine levels, whereas minocycline had more broad effects. These data suggest that use of neuroprotective agents such as TRAM-34 and minocycline should be further explored as a means of reducing the risk of acute DKA-related brain injury. Administration of these agents for 1 week after DKA recovery did not improve long-term neuroinflammation resulting from DKA, however, investigation of more prolonged treatment is necessary before it can be determined whether neuroprotective interventions might have a role in preventing chronic neuroinflammation and cognitive decline resulting from DKA.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the Animal Use and Care Committee at University of California Davis (protocol# 21574).
Acknowledgments
The authors thank Boram Lee for helping them to optimize the procedures for brain tissue lysate processing. The authors also thank Dr. Yi-Je Chen for his help in conducting the small animal procedures. The authors also thank Dr. JoAnn Yee for the helpful assistance in conducting the multiplex assays for these studies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors NG obtained funding, contributed to study design, supervised data collection and data analysis, drafted and revised the manuscript. SC contributed to study design, participated in data collection, assisted with data analysis, reviewed and revised the manuscript. JW contributed to study design, participated in data collection, reviewed and revised the manuscript. LZ contributed to study design, participated in data collection, reviewed and revised the manuscript. HW contributed to study design, assisted in supervising data collection, reviewed and revised the manuscript. DT contributed to study design, analyzed the data in collaboration with NG, reviewed and revised the manuscript. MEO contributed to study design, assisted in supervising data collection, assisted with data analysis, reviewed and revised the manuscript. NG accepts full responsibility as guarantor, for the work and conduct of the study, had access to the data and controlled the decision to publish.
Funding These studies were supported by American Diabetes Association basic science grant 1-17-IBS-186 (to NG).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.