Article Text

Abstract
Introduction Adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate (NPC43) is a recently identified small, non-peptidyl molecule which restores normal insulin signaling in a mouse model of type 2 diabetes (Lan et al). The present study investigated the ability of NPC43 as an oral and injectable insulin-replacing agent to activate insulin receptor (INSR) and counter hyperglycemia in streptozotocin (STZ)-induced type 1 diabetic (T1D) mice.
Research design and methods In this study, STZ was intraperitoneally injected into wild-type mice to induce hyperglycemia and hypoinsulinemia, the main features of T1D. These STZ-induced T1D mice were given NPC43 orally or intraperitoneally and blood glucose levels were measured using a glucometer. Protein levels of phosphorylated and total Insrβ, protein kinase B (Akt) and AS160 (critical for glucose uptake) in the skeletal muscle and liver of STZ-induced T1D mice following oral NPC43 treatment were determined by western blot analysis. In addition, hepatic expression of activated Insr in STZ-induced T1D mice after intraperitoneal NPC43 treatment was measured by ELISA. Student’s t-test was used for statistical analysis.
Results Oral administration of NPC43 at a dose of 5.4 or 10.8 mg/kg body weight (mpk) effectively lowered blood glucose levels in STZ-induced T1D mice at ≥1 hour post-treatment and the glucose-lowering activity of oral NPC43 persisted for 5 hours. Blood glucose levels were also reduced in STZ-induced T1D mice following intraperitoneal NPC43 (5.4 mpk) treatment. Protein levels of phosphorylated Insrβ, Akt and AS160 were significantly increased in the skeletal muscle and liver of STZ-induced T1D mice after oral NPC43 (5.4 mpk) treatment. In addition, activation of hepatic Insr was observed in STZ-induced T1D mice following intraperitoneal NPC43 (5.4 mpk) treatment.
Conclusions We conclude that NPC43 is a de facto fast-acting oral and injectable insulin mimetic which activates Insr and mitigates hyperglycemia in a mouse model of T1D.
- diabetes mellitus
- experimental
- hyperglycemia
- mice
- pharmacology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
Adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate (NPC43) can restore insulin signaling in a hyperinsulinemic mouse model of type 2 diabetes (Leprdb/db) and is being developed as a treatment for type 2 diabetes.
NPC43 can effectively mimic insulin by activating insulin receptor (INSR) in cultured skeletal muscle and liver cells; it promotes glucose uptake in cultured liver cells and cooperates with insulin to stimulate skeletal muscle glucose uptake.
What are the new findings?
NPC43, when administered by oral gavage and intraperitoneal injection, effectively attenuated hyperglycemia in streptozotocin-induced type 1 diabetic (T1D) mice.
The glucose-lowering activity of oral NPC43 in streptozotocin-induced T1D mice was due to the activation of Insr and its downstream Akt/AS160 signaling for glucose uptake in skeletal muscle and liver.
How might these results change the focus of research or clinical practice?
Our findings suggest that the use of NPC43 may represent an orally effective, affordable and fast-acting insulin replacement therapy for T1D.
Furthermore, it could constitute a unique, non-insulin-based approach for treating both type 1 and type 2 diabetes.
Introduction
Type 1 diabetes (T1D) mellitus is typically associated with T-cell-driven autoimmune destruction of pancreatic β-cells, leading to cessation of insulin production and a state of hyperglycemia.1 Approximately 10% of patients with diabetes have T1D, making it far less common than type 2 diabetes (T2D), which is characterized by general insulin resistance and eventually lower insulin production.2 3 T1D is, however, the form which most commonly affects children and young people. It is also associated with lower life expectancy than patients with T2D.4 5 Taken together, these factors make T1D a high priority research area for the development of new treatment options.
Insulin injection is essential for the treatment of T1D, but it is also a mainstay of therapy for patients with T2D, especially when the pancreas undergoes progressive β-cell failure. The last decade has seen a rise in insulin costs to unaffordable levels for many patients with diabetes.6 7 Therefore, a safe and effective molecule which can be orally administered to activate insulin receptor (INSR) in an insulin-independent manner would represent an important advancement in general diabetes management.
NPC43 (adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate) is a small, non-peptidyl compound that has recently been identified as an orally effective agent in restoring normal Insr signaling in a hyperglycemic, insulin-resistant mouse model of T2D. Additionally, in cultured liver and skeletal muscle cells, NPC43 mimics insulin to directly activate INSR and its downstream protein kinase B (Akt)/AS160 signaling pathway, which actuates glucose uptake. NPC43 also cooperates with insulin to stimulate skeletal muscle glucose uptake and attenuates hepatic G6pc-driven gluconeogenesis.8
Therefore, it is important to establish if NPC43 can similarly activate INSR and ameliorate hyperglycemia in T1D. In this report, streptozotocin (STZ)-induced T1D mice were administered NPC43 orally and intraperitoneally. Following oral or intraperitoneal NPC43 treatments, blood glucose levels and protein levels of activated Insr in the skeletal muscle and/or liver of STZ-induced T1D mice were measured. Our results demonstrate that NPC43 is effective both as an oral preparation and an injectable in activating Insr and countering hyperglycemia in STZ-induced T1D mice. Thus, in this study, NPC43 was shown to be a de facto insulin replacement in vivo and may represent a novel treatment for T1D.
Methods
Animals
Male C57BL/6J mice 4–5 weeks old were purchased from The Jackson Laboratory (Bar Harbor, Maine) and housed in a pathogen-free vivarium with free access to chow and water. Room temperature at the vivarium was kept at approximately 25°C, with a relative humidity of 60% and a 12/12-hour light/dark cycle.
Generation of STZ-induced T1D mice
STZ-induced T1D mice were generated as previously described.9 In brief, STZ (Cat #S0130, Sigma-Aldrich, St Louis, Missouri) was dissolved in 0.05 M sodium citrate buffer and intraperitoneally injected into male C57BL/6J mice (4–5 weeks old) at a dose of 55 mg/kg body weight (mpk) daily for 5 days. After STZ injection, these mice were housed in the pathogen-free vivarium for 14–48 days and blood glucose levels were measured using a glucometer as described in Lan et al.8 Serum insulin levels in these mice at 48 days post-intraperitoneal STZ treatment were also determined using Thermo Fisher Scientific’s Mouse Insulin ELISA-Kit (Cat #EMINS), according to the manufacturer’s protocol.
NPC43 per os treatments and change in blood glucose levels
NPC43 was synthesized as described in Lan et al8 and the purity of NPC43 was verified to be ≥99%, as determined by liquid chromatography-inductively coupled plasma mass spectrometry (LC-ICP-MS). NPC43 was dissolved in 100% dimethylsulfoxide (DMSO) to generate a 126.7 mM stock solution.
At 16–26 days post-intraperitoneal STZ treatment, animals were fasted for 2 hours and then subjected to oral (by oral gavage) treatment with NPC43 (diluting the NPC43 stock solution with physiological saline to administer 5.4 and 10.8 mpk) or its vehicle (1% (v/v) DMSO/saline, referred to as saline). Animals at 48 days post-STZ treatment were also fasted for 2 hours and orally administered with 5.4 mpk NPC43 or 1% (v/v) DMSO/saline. Blood glucose levels in each mouse before oral gavage (0 time point) and post-treatment (at 1–5 hour time points) were determined using a glucometer as described previously.8 The change in blood glucose level in individual mice was obtained by subtracting the glucose level right before oral gavage from the blood glucose level at each time-period after oral gavage.
NPC43 intraperitoneal treatments and blood glucose analysis
At 14 days post-intraperitoneal STZ treatment, animals with unfasted blood glucose levels between 500 and 550 mg/dL were fasted overnight and injected intraperitoneally with 5.4 mpk NPC43 or its vehicle (2% (v/v) DMSO/saline). At 1, 2 and 3 hours post-NPC43 injection, blood glucose levels in these mice, which had free access to water but not chow, were measured as described previously.8
Western blot analysis
Forty-eight days post-STZ induction, liver tissue was collected for protein analysis from saline-treated and NPC43-treated mice, 3 hours after oral administration. Separately, STZ-induced T1D mice at 48 days post-STZ treatment were also fasted for 2 hours and orally administered with saline or NPC43 (5.4 mpk) for 5 min prior to harvesting skeletal muscle (gastrocnemius). Skeletal muscle protein (75 µg/mouse) or liver protein (100 µg/mouse) was subjected to western blot analysis of Insr signaling molecules, as described previously.8 Primary antibodies against phosphorylated Insrβ (pInsrβ) at Y1146 (Cat #3021), Insrβ (Cat #3025), phosphorylated Akt (pAkt) at T308 (Cat #2965), Akt (Cat #4691), phosphorylated AS160 (pAS160) at S588 (Cat #8730), AS160 (Cat #2670), and Gapdh (Cat #5174) were purchased from Cell Signaling Technology (Danvers, Massachusetts). Protein band densities in the western blots were determined using the National Institutes of Health (NIH) ImageJ software10 to obtain the ratio of phosphorylated form to total protein level of the respective Insr signaling molecules.
ELISA of hepatic pInsrβ at Y1146
Liver tissues were collected from overnight-fasted, STZ-induced T1D mice (at 21 days post-STZ treatment) after intraperitoneal treatment with NPC43 (5.4 mpk) or its vehicle (2% (v/v) DMSO/saline) for 90 min. These liver protein extracts (800 µg/mouse) were subjected to ELISA analysis to determine the protein levels of pInsrβ-Y1146 using the PathScan Phospho-Insulin Receptor β (Tyr1146) Sandwich ELISA Kit (Cell Signaling Technology).
Statistical analysis
Where applicable, a Student’s t-test was used to determine the statistical significance of the difference between the control and NPC43-treated groups.
Results and discussion
STZ-induced T1D mouse models have been widely used for the study of new therapeutic agents for T1D.9 We examined blood glucose levels and serum insulin levels in wild-type mice following intraperitoneal STZ treatment to validate the T1D model system. As expected, blood glucose levels were significantly increased in STZ-treated mice at 19 and 48 days post-intraperitoneal STZ treatment (when compared with wild-type mice injected with sodium citrate buffer; figure 1A), while serum insulin levels were almost undetectable in STZ-treated mice at 48 days post-STZ treatment (figure 1B). These results confirmed that STZ treatment elicited hyperglycemia and hypoinsulinemia in wild-type mice. As such, STZ-treated C57BL/6J mice recapitulated the main features of T1D and were deemed to be a suitable model system in which to study the overall effects of NPC43.

Hyperglycemia and hypoinsulinemia in C57BL/6J mice following intraperitoneal treatment with STZ. Male C57BL6 mice (4–5 weeks old) were intraperitoneally (i.p.) injected with 0.05 M sodium citrate buffer (referred to as wild-type group) or 55 mpk STZ (dissolved in citrate buffer) daily for 5 days. At 19 and 48 days post-STZ treatment, animals were fasted for 2 hours and blood samples were collected from mouse tails for (A) glucose analysis using a glucometer. At 48 days post-STZ treatment, mouse serum samples were collected and subjected to (B) ELISA analysis of insulin. Data are presented as mean±SEM for indicated numbers of mice per group, and p values were determined using the Student’s t-test software. BD, barely detected; mpk, mg/kg body weight; STZ, streptozotocin.
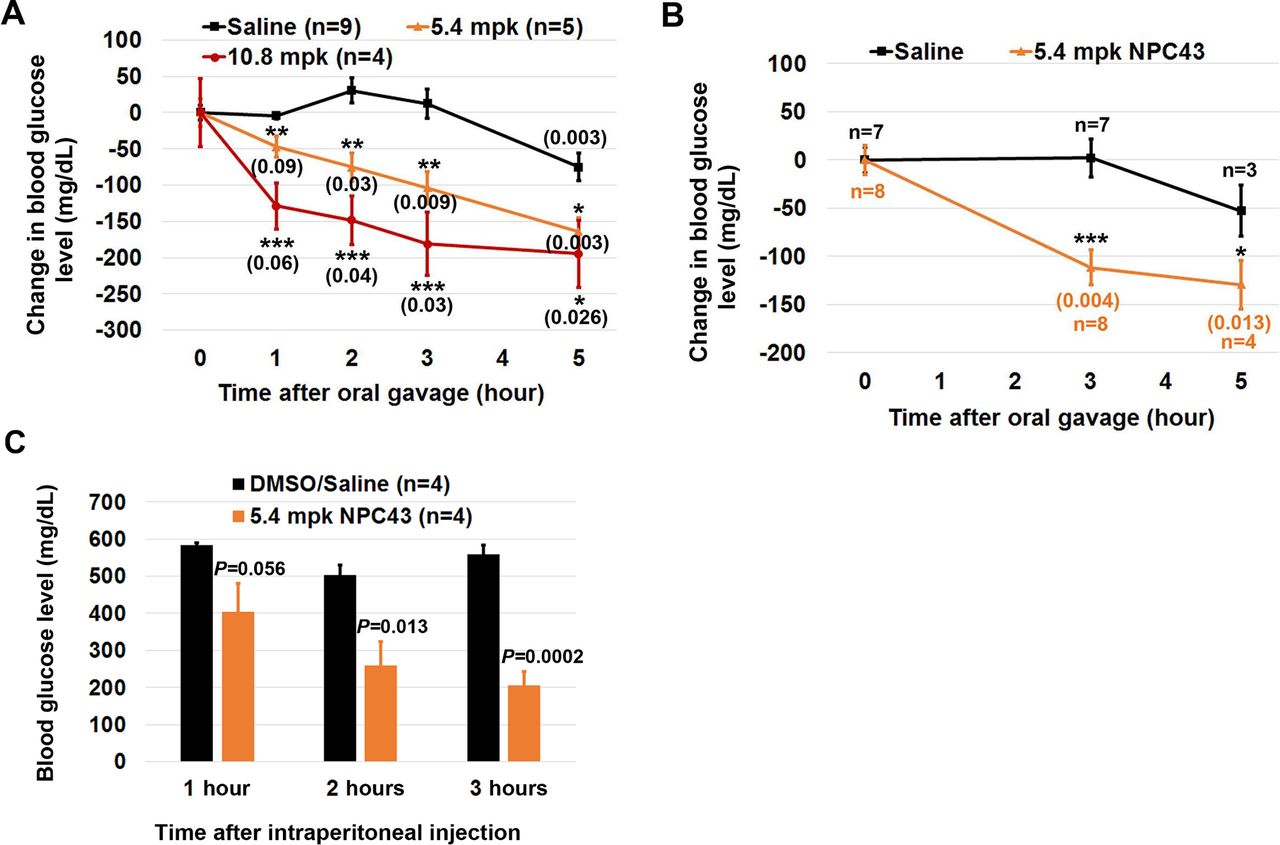
To investigate whether NPC43 can act as an oral insulin replacement to counter hyperglycemia in T1D, STZ-induced T1D mice at 19–26 days post-STZ injection were fasted for 2 hours before oral administration of NPC43 (5.4 and 10.8 mpk) or 1% DMSO/saline (saline) for 0, 1, 2, 3 and 5 hours. Following these treatments, blood glucose levels were determined. As shown in figure 2A, oral saline treatment for 1–3 hours did not affect blood glucose levels even though blood glucose levels in saline-treated mice at 5 hours were slightly decreased when compared with the 0 time point. In contrast, oral NPC43 treatments at both doses in STZ-induced T1D mice caused a gradual decrease in blood glucose levels over the 5-hour test period starting at 1 hour (vs glucose levels at 0 time point) and the glucose-lowering effect of oral NPC43 was maximal at 5 hours (figure 2A). When compared with their control saline counterparts at each time point, the degree of decreased blood glucose levels in T1D mice after oral NPC43 treatment at both tested doses was statistically significant (figure 2A). In addition, a significant decrease in blood glucose levels was also observed in STZ-induced T1D mice at 48 days post-STZ injection (in which serum insulin levels were nearly undetectable; figure 1B) following oral NPC43 (5.4 mpk) treatment for 3 and 5 hours (figure 2B). Together, these results demonstrate that NPC43 is effective, when administered orally, at lowering blood glucose levels in the absence of insulin in a mouse model of T1D.
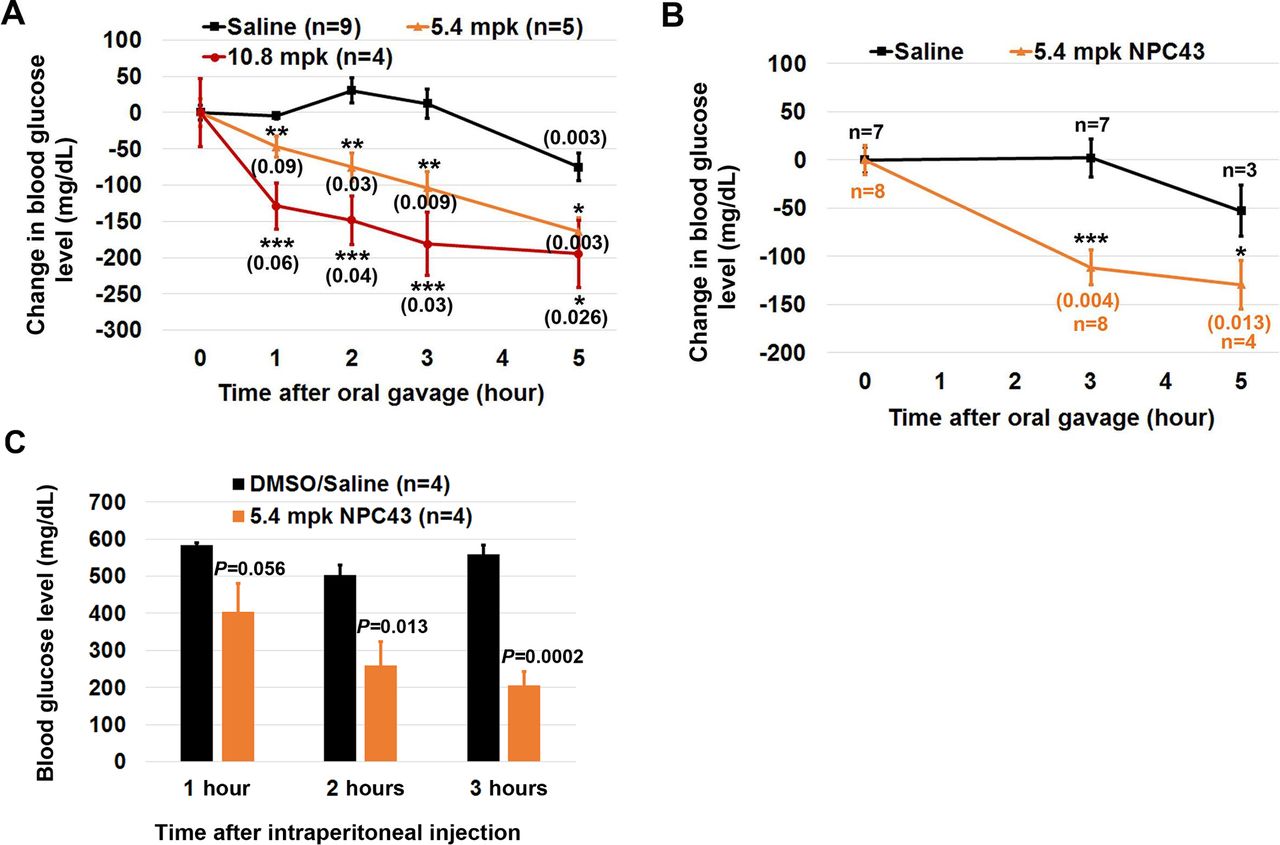
Reduced blood glucose levels in STZ-induced T1D mice after oral and intraperitoneal NPC43 treatments. (A, B) Change in blood glucose levels in STZ-induced mice after oral NPC43 administration. Male C57BL6 mice (4–5 weeks old) were intraperitoneally injected with 55 mpk STZ daily for 5 days. At (A) 19–26 or (B) 48 days post-STZ treatment, STZ-induced T1D mice were fasted for 2 hours and then orally administered with 1% (v/v) dimethylsulfoxide (DMSO)/physiological saline (saline) or indicated dose(s) of NPC43. Blood glucose levels in STZ-induced T1D mice right before and after oral saline or NPC43 treatment at the indicated time points (under fasting conditions but having free access to water) were examined to obtain the change in blood glucose level in each mouse. Mean±SEM, *p<0.05, **p<0.01, ***p<0.001 versus the saline group at the same time point (Student’s t-test). The numbers in parenthesis are p values versus each treatment group before oral gavage (0 time point) (Student’s t-test). After blood glucose analysis in (B), liver tissues of STZ-induced T1D mice at 3 hours post saline or NPC43 treatments (four mice per group) were collected for protein analysis of Insr/Akt/AS160 signaling shown in figure 4. (C) Reduced blood glucose levels in STZ-induced T1D mice after intraperitoneal treatment with NPC43. STZ-induced T1D mice (at 14 days post-STZ treatment) with unfasted blood glucose levels between 500 and 550 mg/dL were fasted overnight and intraperitoneally injected with NPC43 (5.4 mpk) or 2% (v/v) DMSO/saline. At 1, 2 and 3 hours post-intraperitoneal NPC43 injection, blood glucose levels in these mice were measured using the glucometer. Data are presented as mean±SEM of four mice per group. P value (the NPC43-treated group vs the DMSO/saline group at the same time point) was determined by performing Student’s t-test. mpk, mg/kg body weight; NPC43, adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate; STZ, streptozotocin; T1D, type 1 diabetes.
In addition, we also investigated whether acute intraperitoneal treatment with NPC43 elicited a similar antihyperglycemic effect to oral NPC43 administration in STZ-induced T1D mice. As shown in figure 2C, intraperitoneal injection of 5.4 mpk NPC43 into overnight-fasted, STZ-induced T1D mice caused a strong tendency of decreased blood glucose levels at 1 hour post-NPC43 treatment, and more importantly a significant decrease in blood glucose levels at 2 and 3 hours post-NPC43 treatment (figure 2C). Therefore, these results demonstrate that intraperitoneally administered NPC43 can effectively attenuate hyperglycemia in T1D mice in a fashion similar to orally administered NPC43 (figure 2A,B).
It was noted that there was a slight decrease in blood glucose levels in STZ-induced T1D mice following oral DMSO/saline administration at the 5-hour time point (figure 2A,B). This may be due to the small amount of DMSO vehicle since DMSO has been reported to enhance glucose uptake in adipocytes, inhibit spontaneous diabetes in non-obese mice and reduce blood glucose levels in a rat model of polycystic ovarian syndrome.11–13 As a result of this, the persistence of NPC43 action beyond 5 hours was not examined, although simple extrapolation of the line for both dosage levels employed (figure 2A,B) clearly indicates that blood glucose levels would remain lowered beyond the 5-hour mark. In addition, non-fasting glucose levels in response to NPC43 are not presented due to postprandial glucose levels varying in individual animals according to variable food consumption and the possibility that NPC43 absorption might be negatively affected through binding to food particles. Nevertheless, our results (figure 2) clearly demonstrate that both acute oral and intraperitoneal administration of NPC43 at 1–3 hours post-treatment (during which time the DMSO/saline vehicle had zero effect on blood glucose levels) can effectively mitigate hyperglycemia in STZ-induced T1D mice.
INSR and its downstream AKT/AS160 signaling pathway are essential for controlling glucose uptake and maintaining normoglycemia in response to insulin.14 15 Once insulin binds to extracellular INSRα, INSR is activated via phosphorylation of intracellular INSRβ at Y1146, Y1150 and Y1151,16 and elicits the phosphorylation of Akt and AS160.15 17 Phosphorylation of AS160 induces the translocation of cytosolic glucose transporter proteins to the plasma membrane to facilitate glucose uptake from the bloodstream, resulting in a decrease in blood glucose level in vivo. The observed antihyperglycemic activity of oral NPC43 in STZ-induced T1D mice (figure 2) may be attributed to the activation of Insr/Akt/AS160 signaling by NPC43 to enhance glucose utilization by the skeletal muscle and liver. Thus, the protein levels of pInsrβ at Y1146 (the marker for activated Insr),16 pAkt at T308 and pAS160 at S588 in the gastrocnemius and liver of STZ-induced T1D mice, following oral treatment with 5.4 mpk NPC43, were determined by western blot analysis.
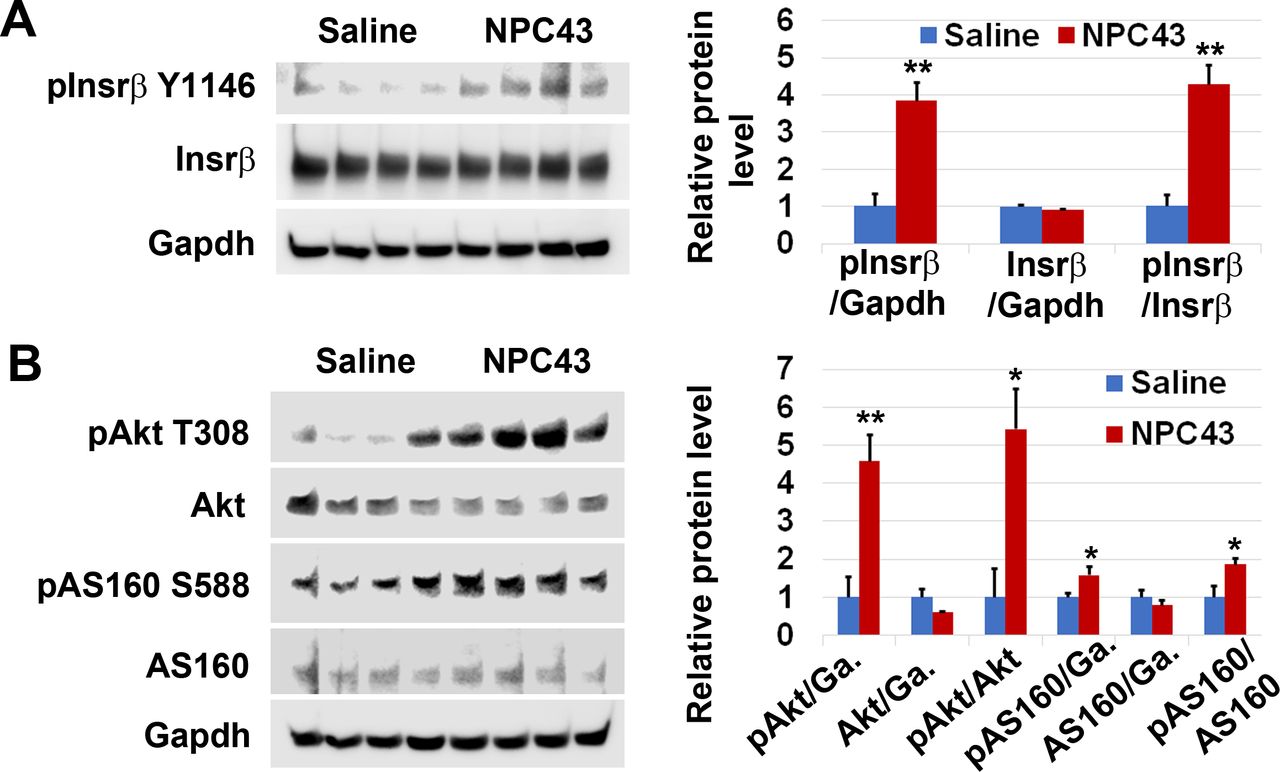
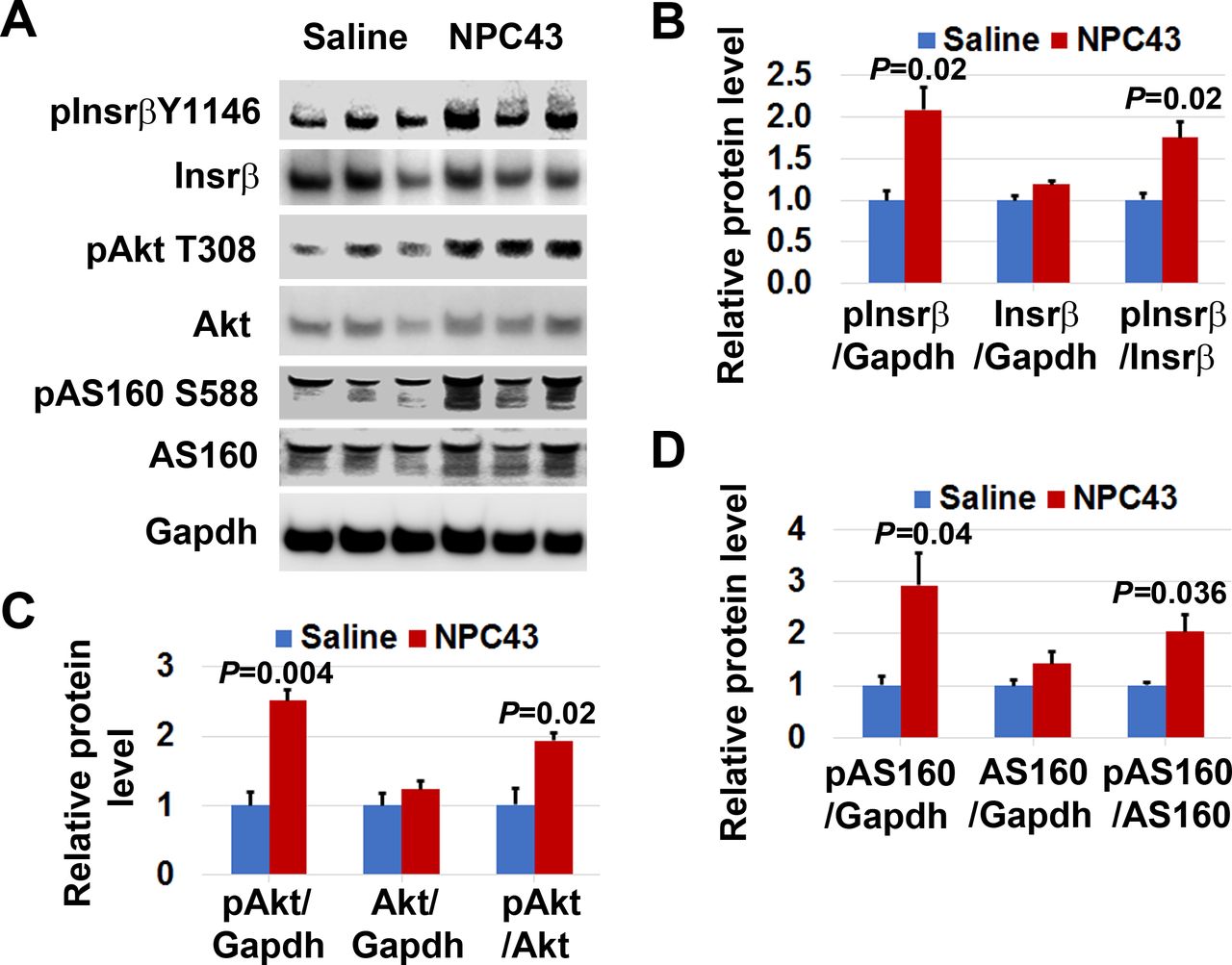
We first examined protein expression associated with Insr/Akt/AS160 signaling in the gastrocnemius of STZ-induced T1D mice after oral treatment with 5.4 mpk NPC43 for 5 min. As shown in figure 3A, protein expression of pInsrβ at Y1146 and pAkt at T308, but not total Insrβ or Akt, was visibly increased in the skeletal muscle of STZ-induced T1D mice after oral NPC43 treatment for just 5 min. Quantitative analysis showed that there was a significant 1.8-fold and 1.9-fold increase in the ratio of pInsrβ and pAkt protein levels to total Insrβ and Akt protein levels, respectively, in the skeletal muscle of STZ-induced T1D mice post-NPC43 treatment for 5 min, when compared with saline-treated mice (figure 3B,C). In the skeletal muscle of STZ-induced T1D mice, multiple protein bands were detected by specific monoclonal antibodies against pAS160 at S588 or total AS160 in western blots (figure 3A). We have repeated these experiments twice and observed the same results. Thus, all these protein bands detected by pAS160 or AS160 monoclonal antibodies are most likely AS160 isoforms existing in the skeletal muscle of STZ-induced T1D mice. As shown in figure 3A, there was a visible increase in protein expression of pAS160 at S588, but not total AS160, in the skeletal muscle of STZ-induced T1D mice after oral NPC43 treatment for 5 min. Quantitative analysis of all those detected protein bands showed a significant twofold increase in the ratio of pAS160 at S588 to total AS160 protein levels in the skeletal muscle of NPC43-treated T1D mice, when compared with saline-treated mice (figure 3D). Together, these results demonstrate that NPC43 can very rapidly activate Insr and its downstream signaling molecules, Akt and AS160, in the skeletal muscle of STZ-induced T1D mice.
Activation of Insr and stimulation of Akt and AS160 phosphorylation in the skeletal muscle of STZ-induced T1D mice by NPC43. Male C57BL6 mice (4–5 weeks old) were intraperitoneally injected with 55 mpk STZ daily for 5 days. At 48 days post-STZ treatment, animals were fasted 2 hours and then orally administered with 1% (v/v) dimethylsulfoxide (DMSO)/physiological saline (saline, n=3) or NPC43 (5.4 mpk, n=3) for 5 min. Gastrocnemius from these mice was collected and subjected to (A) western blot analysis by the indicated antibodies (using 75 µg protein/mouse). (B–D) Quantitative changes in protein expression of (B) pInsrβ-Y1146/Insrβ, (C) pAkt-T308/Akt, and (D) pAS160-S588/AS160 in the skeletal muscle of STZ-induced T1D mice after acute oral administration of NPC43, as determined by the western blot analysis shown in (A). Protein band densities in western blots were determined using National Institutes of Health (NIH) ImageJ software. For pAS160-S588 and AS160 protein expression, all protein bands detected by their specific monoclonal antibodies in western blots were quantitated. Data are presented as mean±SEM of three mice per group. P value (the NPC43-treated group vs the control saline group) was determined by performing Student’s t-test. Insr, insulin receptor; mpk, mg/kg body weight; NPC43, adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate; STZ, streptozotocin; T1D, type 1 diabetes.
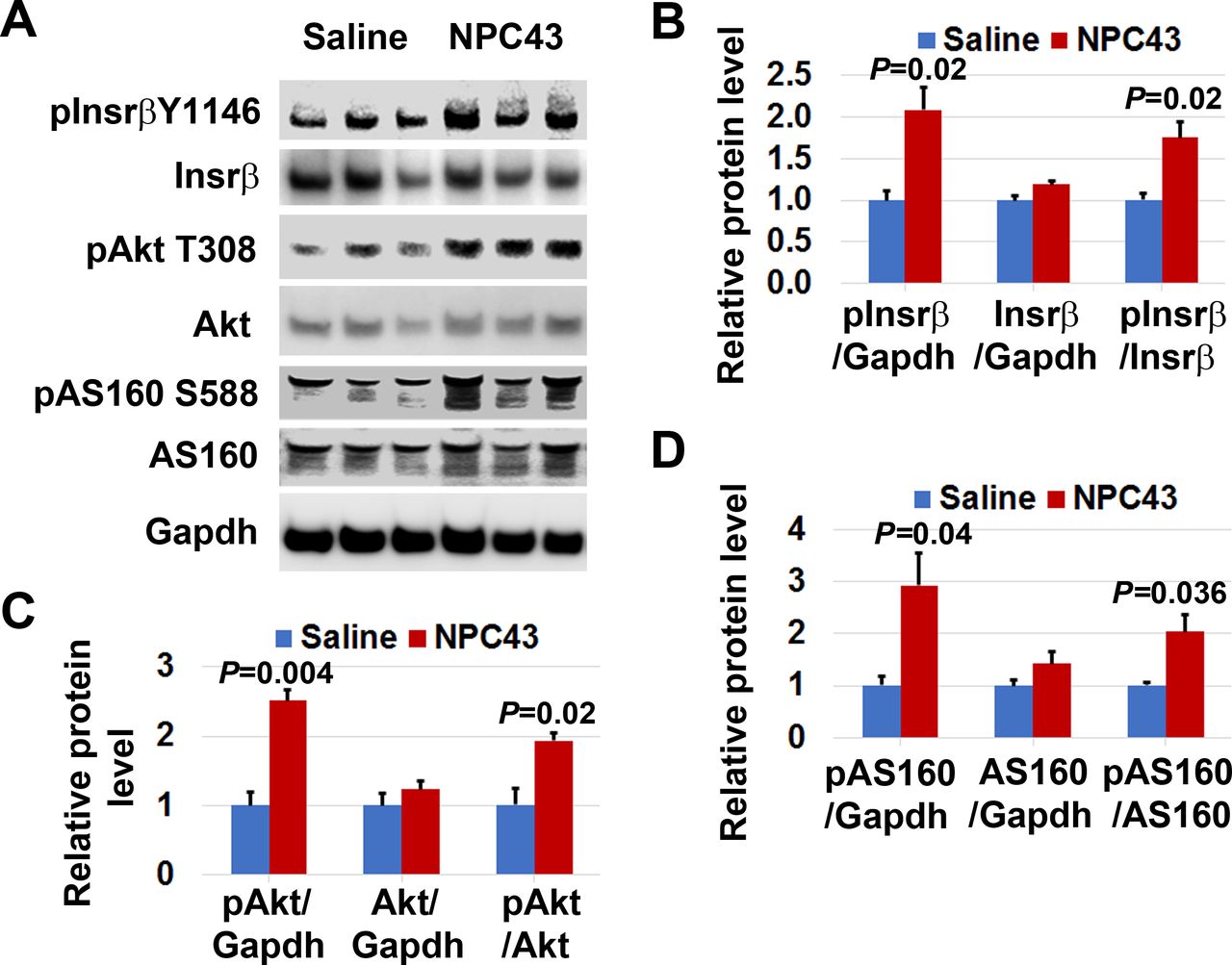
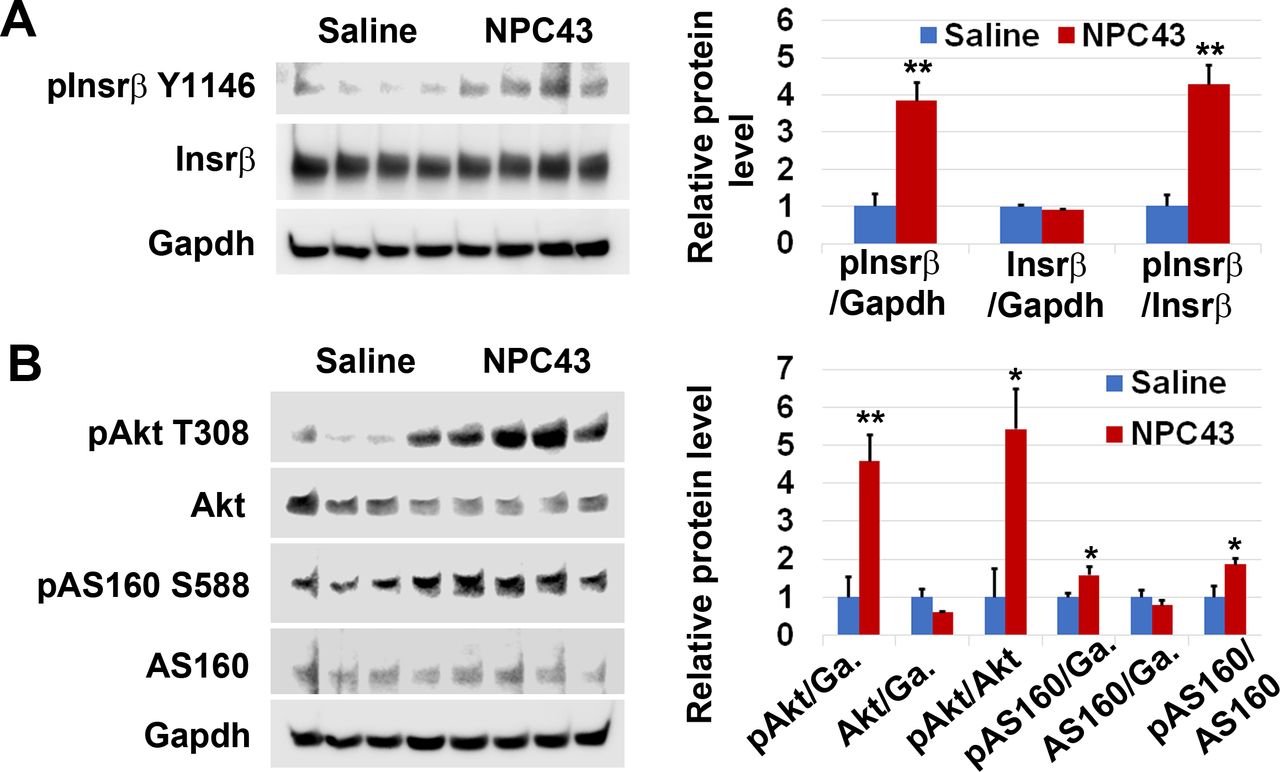
Similarly, we also examined the phosphorylation status of Insrβ at Y1146, pAkt at T308 and pAS160 at S588 in the liver of STZ-induced T1D mice after oral treatment with 5.4 mpk NPC43 for 3 hours, at which time blood glucose levels were significantly decreased (figure 2B). As shown in figure 4A, protein levels of pInsrβ at Y1146, but not total Insrβ, were significantly increased (~4.2-fold increase vs the control saline group) in the liver of NPC43-treated T1D mice. In addition, a significant increase in the ratio of pAkt at T308 to total Akt protein levels (~5.4-fold increase), and pAS160 at S588 to total AS160 protein levels (~1.9-fold increase), was also observed in the liver of STZ-induced T1D mice after NPC43 treatment for 3 hours, when compared with saline-treated mice (figure 4B). These results indicate that NPC43 can mimic insulin to activate Insr and its downstream Akt/AS160 signaling in the liver cells of STZ-induced T1D mice.
Activation of Insr and stimulation of Akt and AS160 phosphorylation in the liver of STZ-induced T1D mice by NPC43. Male C57BL6 mice (4–5 weeks old) were intraperitoneally injected with 55 mpk STZ daily for 5 days. At 48 days post-STZ treatment, animals were fasted 2 hours and then orally administered with 1% (v/v) dimethylsulfoxide (DMSO)/physiological saline (saline, n=4) or NPC43 (5.4 mpk, n=4) for 3 hours. Blood glucose levels in these mice after saline and NPC43 treatments are shown in figure 2B. Liver tissue from these mice was collected and subjected to western blot analysis of (A) activated Insr and (B) phosphorylated Akt and AS160 with the indicated antibodies (using 100 µg liver protein/mouse). Protein band densities in western blots shown in the left panels of A–B were determined using National Institutes of Health (NIH) ImageJ software, and data are presented as mean±SEM of four mice per group in the right bar graphs. In the bar graph shown in (B), ‘Ga.’ means Gapdh. *P<0.05, **P<0.01 versus the control saline group (Student’s t-test). Insr, insulin receptor; mpk, mg/kg body weight; NPC43, adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate; STZ, streptozotocin; T1D, type 1 diabetes.
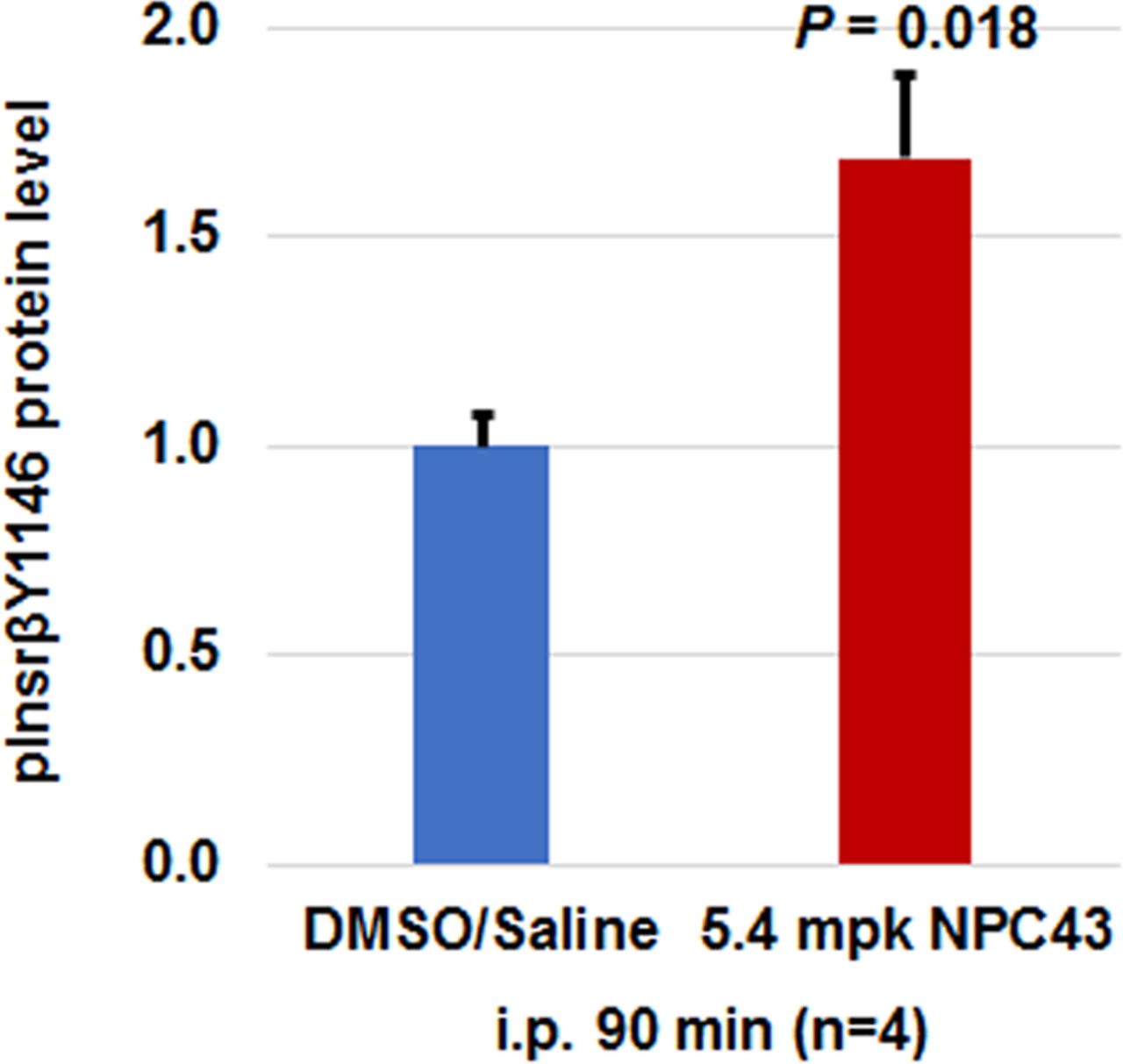
Finally, we investigated whether acute intraperitoneal treatment with NPC43 can activate Insr in the liver, in a similar fashion to oral NPC43 administration in STZ-induced T1D mice. ELISA analysis was performed to examine the protein levels of activated Insr (ie, pInsrβ at Y1146) in the liver of STZ-induced mice after intraperitoneal NPC43 treatment for 90 min. As shown in figure 5, protein levels of pInsrβ at Y1146 were significantly increased in STZ-induced T1D mouse liver after intraperitoneal NPC43 treatment for 90 min when compared with control DMSO/saline-treated mice. These results demonstrate that intraperitoneally administered NPC43 can activate Insr in the liver in a fashion similar to orally administered NPC43 (figure 4A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Activation of hepatic Insr in STZ-induced T1D mice after intraperitoneal (i.p.) treatment with NPC43. STZ-induced T1D mice (at 21 days post-STZ treatment) were fasted overnight and intraperitoneally injected with 2% (v/v) dimethylsulfoxide (DMSO)/saline or NPC43 (5.4 mpk) for 90 min. Liver tissues (using 800 µg liver protein/mouse) were collected and subjected to ELISA analysis of pInsrβ-Y1146. Data are presented as mean±SEM of four mice per group in the bar graphs, and p value (the NPC43-treated group vs the control DMSO/saline group) was determined by performing Student’s t-test. Insr, insulin receptor; mpk, mg/kg body weight; NPC43, adenosine, 5’-Se-methyl-5’-seleno-,2’,3’-diacetate; STZ, streptozotocin; T1D, type 1 diabetes.
These in vivo findings (figures 3–5) are consistent with the activation of Insr/Akt/AS160 signaling previously observed in cultured skeletal muscle and liver cells after NPC43 treatment.8 NPC43 can stimulate glucose uptake in cultured liver cells and cooperate with insulin to enhance glucose uptake in cultured skeletal muscle cells.8 Thus, the mechanism of NPC43 in lowering blood glucose levels in T1D mice (figure 2) is due, in part at least, to enhanced skeletal muscle and hepatic glucose uptake following activation of Insr/Akt/AS160 signaling by NPC43 (figures 3–5). Restoration of insulin signaling and glucose import implies that glycolysis will have a role to play in reinstating normoglycemia. However, it should also be pointed out that we previously have demonstrated that NPC43 inhibits hepatic gluconeogenesis in T2D mice, through Insr/Akt activation and G6pc suppression.8 In the present study, we again observed NPC43-driven Insr/Akt activation in the liver (figures 4–5) and so would surmise that suppressed gluconeogenesis may contribute to reducing blood glucose in T1D also.
Conclusions
In this report, our results demonstrated that acute oral or intraperitoneal administration of NPC43 activates Insr and mitigates hyperglycemia in a mouse model of T1D, indicating that NPC43 is a de facto insulin mimetic in vivo. NPC43 is fast-acting, as indicated by activation of skeletal muscle Insr/Akt/AS160 signaling at just 5 min post oral administration (figure 3), and decreased blood glucose concentration at ≥1 hour post oral or intraperitoneal treatment (figure 2). Our results indicate that the glucose-lowering activity persists for at least 5 hours (figure 2A,B), but the exact pharmacodynamics of NPC43 remain to be determined. Together with our previous studies which demonstrate the efficacy of NPC43 in cell culture and a mouse model of severe insulin resistance/T2D,8 we believe that NPC43 represents a very significant advance in the search for effective and affordable alternatives to insulin. Aside from applications in diabetes management, NPC43 constitutes a promising candidate compound for assessment in the treatment and control of a much broader range of disorders and risk factors associated with insulin resistance syndrome.
Acknowledgments
We thank Ms Jeannie Francis for administrative support.
References
Footnotes
Z-JL and RP contributed equally.
Contributors Z-JL and RP designed the experiments, performed the data analysis, and wrote the manuscript. AY and TRY synthesized NPC43. Z-JL, ZL, LN, XL, HK, KE, RX and RG performed the experiments.
Funding This work was supported by Alltech’s internal life science research program.
Competing interests ZL and XL have no competing interests. All other authors are current or former employees of Alltech. Results in figure 2C have been submitted in the patent application (PCT/US18/30371) filed by Alltech.
Patient consent for publication Not required.
Ethics approval All animal experiments were preapproved by Alltech and performed according to the US National Institutes of Health’s Animal Welfare guidelines. Humane treatment of all research animals was assured. This article does not contain any studies with human participants. All authors confirm that ethical principles were followed in the experiments of this study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article.